 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable training of graph convolutional neural networks for fast and accurate predictions of HOMO-LUMO gap in molecules
Jul 22, 2022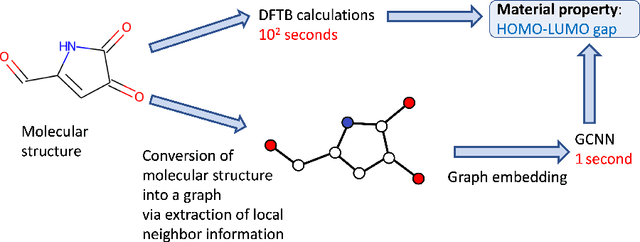

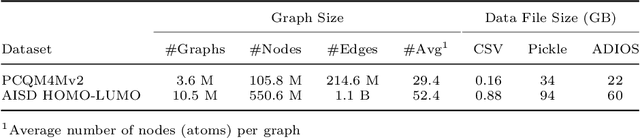
Graph Convolutional Neural Network (GCNN) is a popular class of deep learning (DL) models in material science to predict material properties from the graph representation of molecular structures. Training an accurate and comprehensive GCNN surrogate for molecular design requires large-scale graph datasets and is usually a time-consuming process. Recent advances in GPUs and distributed computing open a path to reduce the computational cost for GCNN training effectively. However, efficient utilization of high performance computing (HPC) resources for training requires simultaneously optimizing large-scale data management and scalable stochastic batched optimization techniques. In this work, we focus on building GCNN models on HPC systems to predict material properties of millions of molecules. We use HydraGNN, our in-house library for large-scale GCNN training, leveraging distributed data parallelism in PyTorch. We use ADIOS, a high-performance data management framework for efficient storage and reading of large molecular graph data. We perform parallel training on two open-source large-scale graph datasets to build a GCNN predictor for an important quantum property known as the HOMO-LUMO gap. We measure the scalability, accuracy, and convergence of our approach on two DOE supercomputers: the Summit supercomputer at the Oak Ridge Leadership Computing Facility (OLCF) and the Perlmutter system at the National Energy Research Scientific Computing Center (NERSC). We present our experimental results with HydraGNN showing i) reduction of data loading time up to 4.2 times compared with a conventional method and ii) linear scaling performance for training up to 1,024 GPUs on both Summit and Perlmutter.