 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgefastHDMI: Fast Mutual Information Estimation for High-Dimensional Data
Oct 14, 2024



In this paper, we introduce fastHDMI, a Python package designed for efficient variable screening in high-dimensional datasets, particularly neuroimaging data. This work pioneers the application of three mutual information estimation methods for neuroimaging variable selection, a novel approach implemented via fastHDMI. These advancements enhance our ability to analyze the complex structures of neuroimaging datasets, providing improved tools for variable selection in high-dimensional spaces. Using the preprocessed ABIDE dataset, we evaluate the performance of these methods through extensive simulations. The tests cover a range of conditions, including linear and nonlinear associations, as well as continuous and binary outcomes. Our results highlight the superiority of the FFTKDE-based mutual information estimation for feature screening in continuous nonlinear outcomes, while binning-based methods outperform others for binary outcomes with nonlinear probability preimages. For linear simulations, both Pearson correlation and FFTKDE-based methods show comparable performance for continuous outcomes, while Pearson excels in binary outcomes with linear probability preimages. A comprehensive case study using the ABIDE dataset further demonstrates fastHDMI's practical utility, showcasing the predictive power of models built from variables selected using our screening techniques. This research affirms the computational efficiency and methodological strength of fastHDMI, significantly enriching the toolkit available for neuroimaging analysis.
Benchmarking missing-values approaches for predictive models on health databases
Feb 17, 2022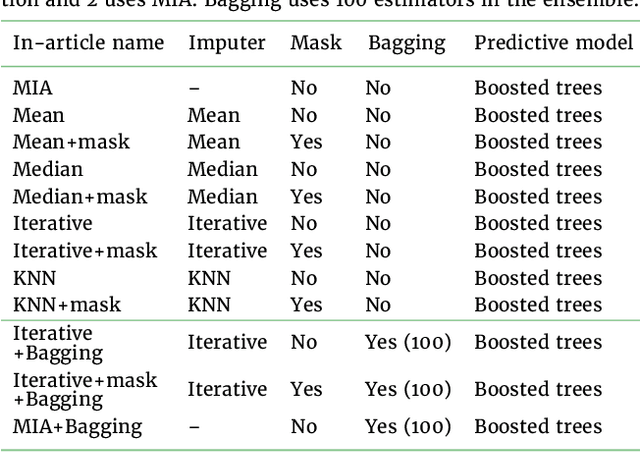
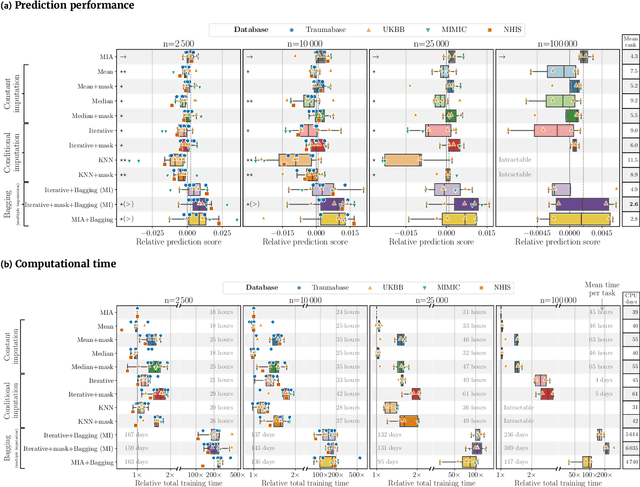
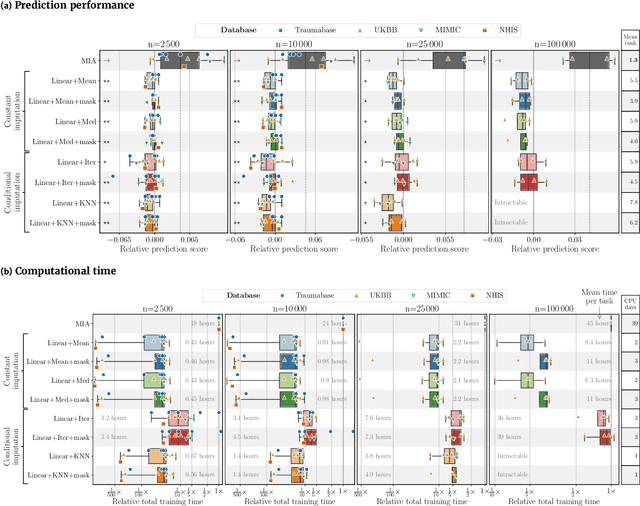
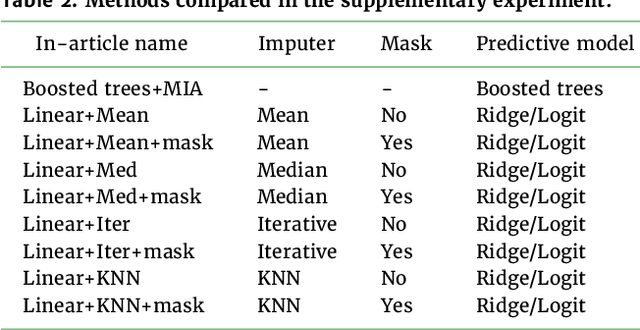
BACKGROUND: As databases grow larger, it becomes harder to fully control their collection, and they frequently come with missing values: incomplete observations. These large databases are well suited to train machine-learning models, for instance for forecasting or to extract biomarkers in biomedical settings. Such predictive approaches can use discriminative -- rather than generative -- modeling, and thus open the door to new missing-values strategies. Yet existing empirical evaluations of strategies to handle missing values have focused on inferential statistics. RESULTS: Here we conduct a systematic benchmark of missing-values strategies in predictive models with a focus on large health databases: four electronic health record datasets, a population brain imaging one, a health survey and two intensive care ones. Using gradient-boosted trees, we compare native support for missing values with simple and state-of-the-art imputation prior to learning. We investigate prediction accuracy and computational time. For prediction after imputation, we find that adding an indicator to express which values have been imputed is important, suggesting that the data are missing not at random. Elaborate missing values imputation can improve prediction compared to simple strategies but requires longer computational time on large data. Learning trees that model missing values-with missing incorporated attribute-leads to robust, fast, and well-performing predictive modeling. CONCLUSIONS: Native support for missing values in supervised machine learning predicts better than state-of-the-art imputation with much less computational cost. When using imputation, it is important to add indicator columns expressing which values have been imputed.
Preventing dataset shift from breaking machine-learning biomarkers
Jul 21, 2021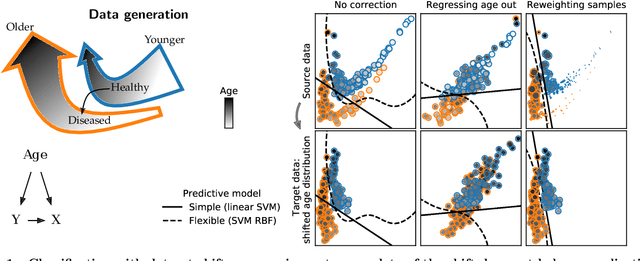
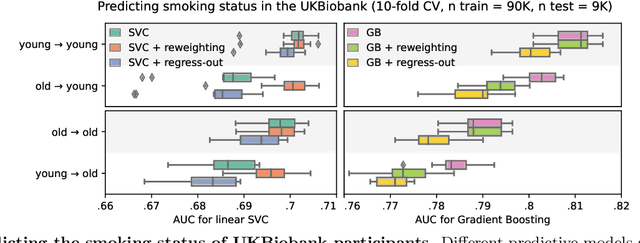
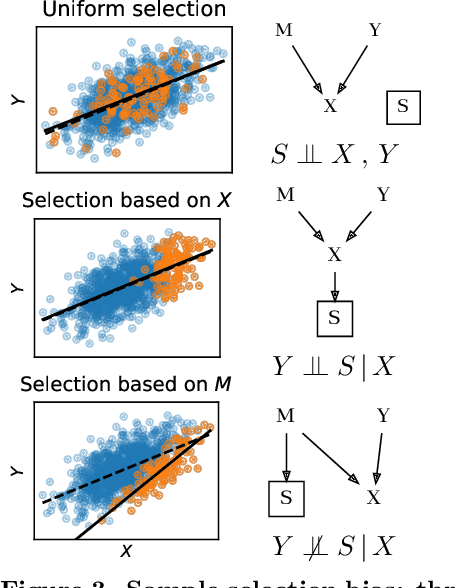
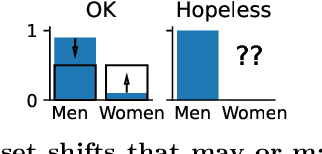
Machine learning brings the hope of finding new biomarkers extracted from cohorts with rich biomedical measurements. A good biomarker is one that gives reliable detection of the corresponding condition. However, biomarkers are often extracted from a cohort that differs from the target population. Such a mismatch, known as a dataset shift, can undermine the application of the biomarker to new individuals. Dataset shifts are frequent in biomedical research, e.g. because of recruitment biases. When a dataset shift occurs, standard machine-learning techniques do not suffice to extract and validate biomarkers. This article provides an overview of when and how dataset shifts breaks machine-learning extracted biomarkers, as well as detection and correction strategies.
An algorithm-based multiple detection influence measure for high dimensional regression using expectile
May 26, 2021


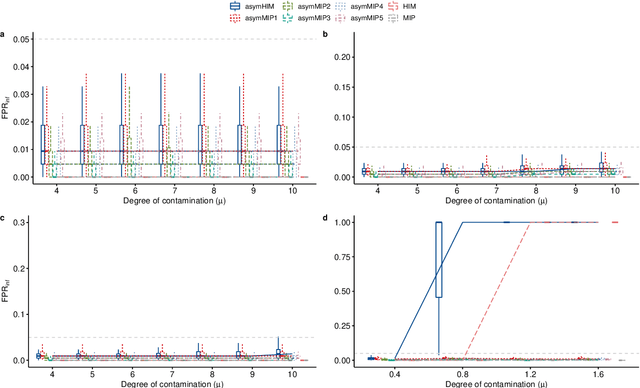
The identification of influential observations is an important part of data analysis that can prevent erroneous conclusions drawn from biased estimators. However, in high dimensional data, this identification is challenging. Classical and recently-developed methods often perform poorly when there are multiple influential observations in the same dataset. In particular, current methods can fail when there is masking several influential observations with similar characteristics, or swamping when the influential observations are near the boundary of the space spanned by well-behaved observations. Therefore, we propose an algorithm-based, multi-step, multiple detection procedure to identify influential observations that addresses current limitations. Our three-step algorithm to identify and capture undesirable variability in the data, $\asymMIP,$ is based on two complementary statistics, inspired by asymmetric correlations, and built on expectiles. Simulations demonstrate higher detection power than competing methods. Use of the resulting asymptotic distribution leads to detection of influential observations without the need for computationally demanding procedures such as the bootstrap. The application of our method to the Autism Brain Imaging Data Exchange neuroimaging dataset resulted in a more balanced and accurate prediction of brain maturity based on cortical thickness. See our GitHub for a free R package that implements our algorithm: \texttt{asymMIP} (\url{github.com/AmBarry/hidetify}).
PyXNAT: XNAT in Python
Jan 29, 2013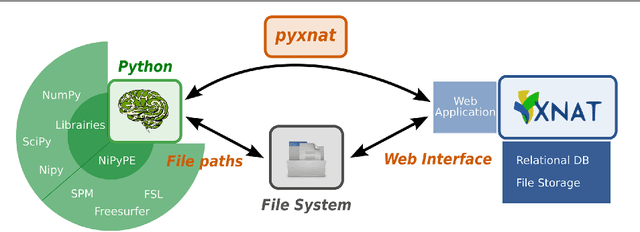


As neuroimaging databases grow in size and complexity, the time researchers spend investigating and managing the data increases to the expense of data analysis. As a result, investigators rely more and more heavily on scripting using high-level languages to automate data management and processing tasks. For this, a structured and programmatic access to the data store is necessary. Web services are a first step toward this goal. They however lack in functionality and ease of use because they provide only low level interfaces to databases. We introduce here PyXNAT, a Python module that interacts with The Extensible Neuroimaging Archive Toolkit (XNAT) through native Python calls across multiple operating systems. The choice of Python enables PyXNAT to expose the XNAT Web Services and unify their features with a higher level and more expressive language. PyXNAT provides XNAT users direct access to all the scientific packages in Python. Finally PyXNAT aims to be efficient and easy to use, both as a backend library to build XNAT clients and as an alternative frontend from the command line.
Improving accuracy and power with transfer learning using a meta-analytic database
Sep 28, 2012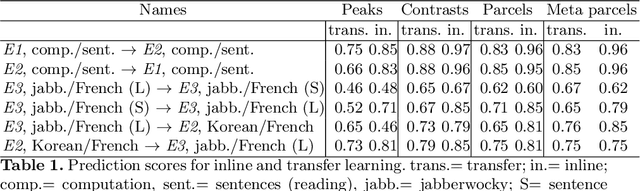
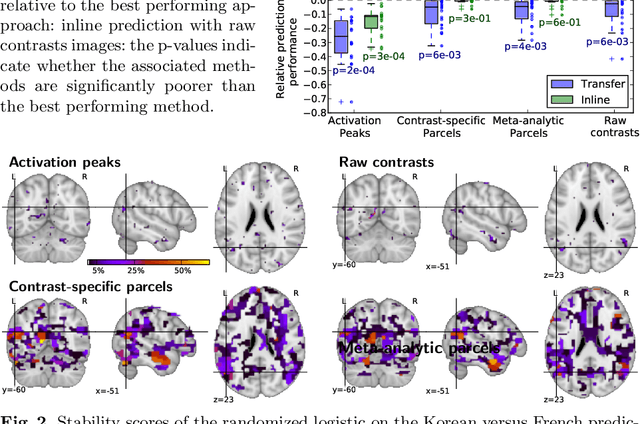

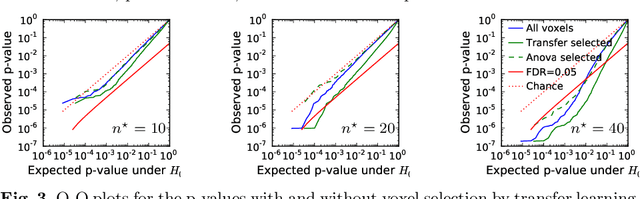
Typical cohorts in brain imaging studies are not large enough for systematic testing of all the information contained in the images. To build testable working hypotheses, investigators thus rely on analysis of previous work, sometimes formalized in a so-called meta-analysis. In brain imaging, this approach underlies the specification of regions of interest (ROIs) that are usually selected on the basis of the coordinates of previously detected effects. In this paper, we propose to use a database of images, rather than coordinates, and frame the problem as transfer learning: learning a discriminant model on a reference task to apply it to a different but related new task. To facilitate statistical analysis of small cohorts, we use a sparse discriminant model that selects predictive voxels on the reference task and thus provides a principled procedure to define ROIs. The benefits of our approach are twofold. First it uses the reference database for prediction, i.e. to provide potential biomarkers in a clinical setting. Second it increases statistical power on the new task. We demonstrate on a set of 18 pairs of functional MRI experimental conditions that our approach gives good prediction. In addition, on a specific transfer situation involving different scanners at different locations, we show that voxel selection based on transfer learning leads to higher detection power on small cohorts.