 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMAMMAL -- Molecular Aligned Multi-Modal Architecture and Language
Oct 28, 2024Drug discovery typically consists of multiple steps, including identifying a target protein key to a disease's etiology, validating that interacting with this target could prevent symptoms or cure the disease, discovering a small molecule or biologic therapeutic to interact with it, and optimizing the candidate molecule through a complex landscape of required properties. Drug discovery related tasks often involve prediction and generation while considering multiple entities that potentially interact, which poses a challenge for typical AI models. For this purpose we present MAMMAL - Molecular Aligned Multi-Modal Architecture and Language - a method that we applied to create a versatile multi-task foundation model ibm/biomed.omics.bl.sm.ma-ted-458m that learns from large-scale biological datasets (2 billion samples) across diverse modalities, including proteins, small molecules, and genes. We introduce a prompt syntax that supports a wide range of classification, regression, and generation tasks. It allows combining different modalities and entity types as inputs and/or outputs. Our model handles combinations of tokens and scalars and enables the generation of small molecules and proteins, property prediction, and transcriptomic lab test predictions. We evaluated the model on 11 diverse downstream tasks spanning different steps within a typical drug discovery pipeline, where it reaches new SOTA in 9 tasks and is comparable to SOTA in 2 tasks. This performance is achieved while using a unified architecture serving all tasks, in contrast to the original SOTA performance achieved using tailored architectures. The model code and pretrained weights are publicly available at https://github.com/BiomedSciAI/biomed-multi-alignment and https://huggingface.co/ibm/biomed.omics.bl.sm.ma-ted-458m.
Never Train from Scratch: Fair Comparison of Long-Sequence Models Requires Data-Driven Priors
Oct 04, 2023Modeling long-range dependencies across sequences is a longstanding goal in machine learning and has led to architectures, such as state space models, that dramatically outperform Transformers on long sequences. However, these impressive empirical gains have been by and large demonstrated on benchmarks (e.g. Long Range Arena), where models are randomly initialized and trained to predict a target label from an input sequence. In this work, we show that random initialization leads to gross overestimation of the differences between architectures and that pretraining with standard denoising objectives, using $\textit{only the downstream task data}$, leads to dramatic gains across multiple architectures and to very small gaps between Transformers and state space models (SSMs). In stark contrast to prior works, we find vanilla Transformers to match the performance of S4 on Long Range Arena when properly pretrained, and we improve the best reported results of SSMs on the PathX-256 task by 20 absolute points. Subsequently, we analyze the utility of previously-proposed structured parameterizations for SSMs and show they become mostly redundant in the presence of data-driven initialization obtained through pretraining. Our work shows that, when evaluating different architectures on supervised tasks, incorporation of data-driven priors via pretraining is essential for reliable performance estimation, and can be done efficiently.
Graph Neural Networks Use Graphs When They Shouldn't
Sep 08, 2023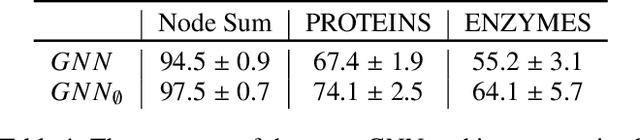
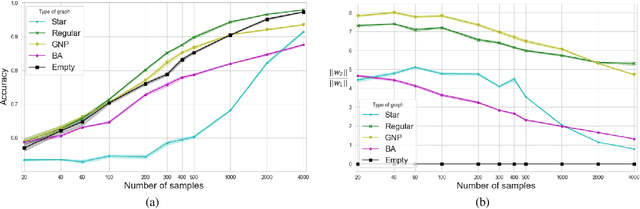
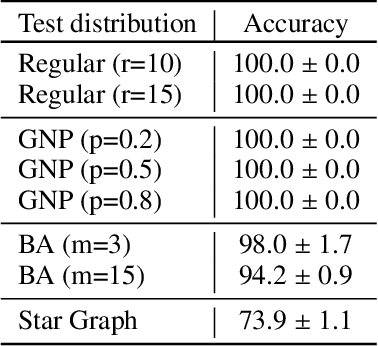
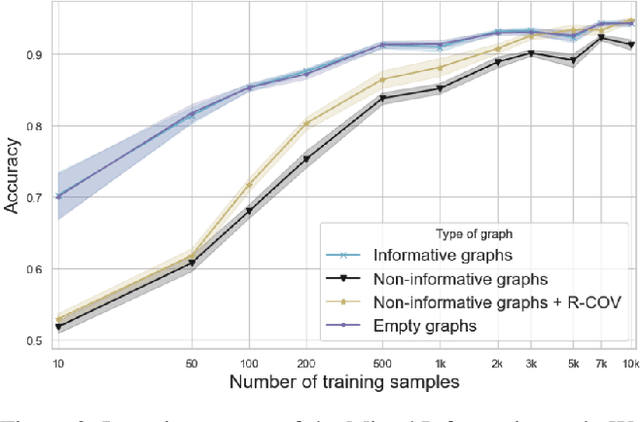
Predictions over graphs play a crucial role in various domains, including social networks, molecular biology, medicine, and more. Graph Neural Networks (GNNs) have emerged as the dominant approach for learning on graph data. Instances of graph labeling problems consist of the graph-structure (i.e., the adjacency matrix), along with node-specific feature vectors. In some cases, this graph-structure is non-informative for the predictive task. For instance, molecular properties such as molar mass depend solely on the constituent atoms (node features), and not on the molecular structure. While GNNs have the ability to ignore the graph-structure in such cases, it is not clear that they will. In this work, we show that GNNs actually tend to overfit the graph-structure in the sense that they use it even when a better solution can be obtained by ignoring it. We examine this phenomenon with respect to different graph distributions and find that regular graphs are more robust to this overfitting. We then provide a theoretical explanation for this phenomenon, via analyzing the implicit bias of gradient-descent-based learning of GNNs in this setting. Finally, based on our empirical and theoretical findings, we propose a graph-editing method to mitigate the tendency of GNNs to overfit graph-structures that should be ignored. We show that this method indeed improves the accuracy of GNNs across multiple benchmarks.
ASOCEM: Automatic Segmentation Of Contaminations in cryo-EM
Jan 18, 2022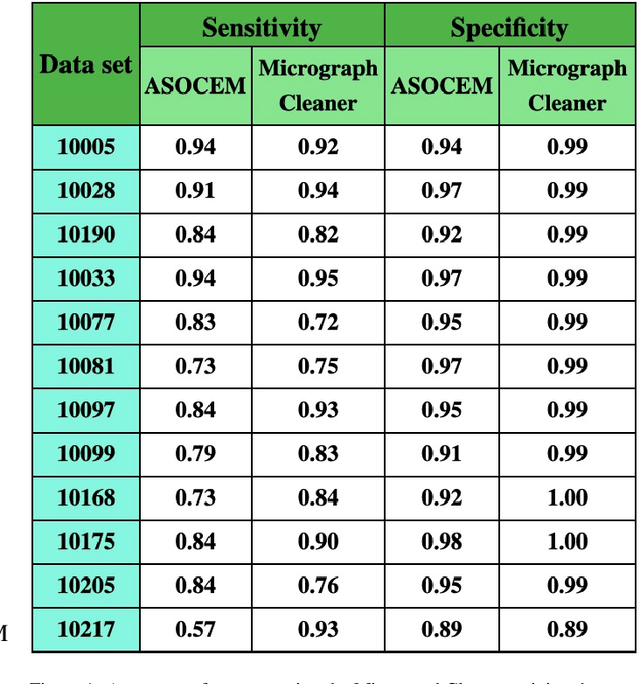

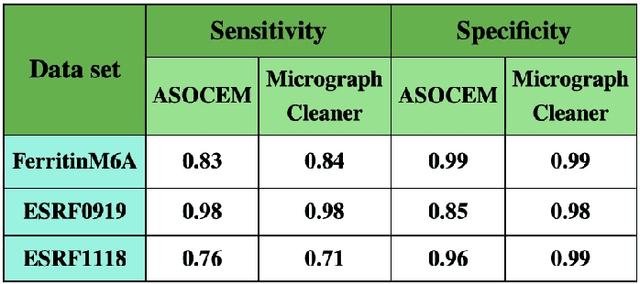

Particle picking is currently a critical step in the cryo-electron microscopy single particle reconstruction pipeline. Contaminations in the acquired micrographs severely degrade the performance of particle pickers, resulting is many ``non-particles'' in the collected stack of particles. In this paper, we present ASOCEM (Automatic Segmentation Of Contaminations in cryo-EM), an automatic method to detect and segment contaminations, which requires as an input only the approximated particle size. In particular, it does not require any parameter tuning nor manual intervention. Our method is based on the observation that the statistical distribution of contaminated regions is different from that of the rest of the micrograph. This nonrestrictive assumption allows to automatically detect various types of contaminations, from the carbon edges of the supporting grid to high contrast blobs of different sizes. We demonstrate the efficiency of our algorithm using various experimental data sets containing various types of contaminations. ASOCEM is integrated as part of the KLT picker \cite{ELDAR2020107473} and is available at \url{https://github.com/ShkolniskyLab/kltpicker2}.