 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Neural Networks Use Graphs When They Shouldn't
Paper and Code
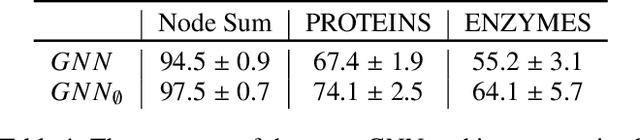
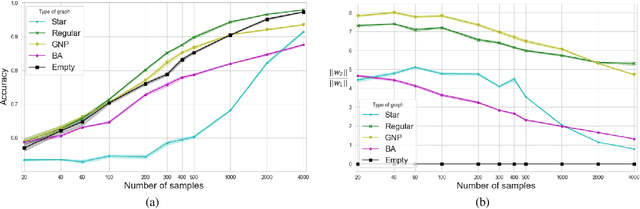
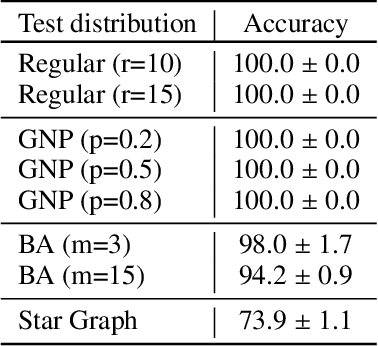
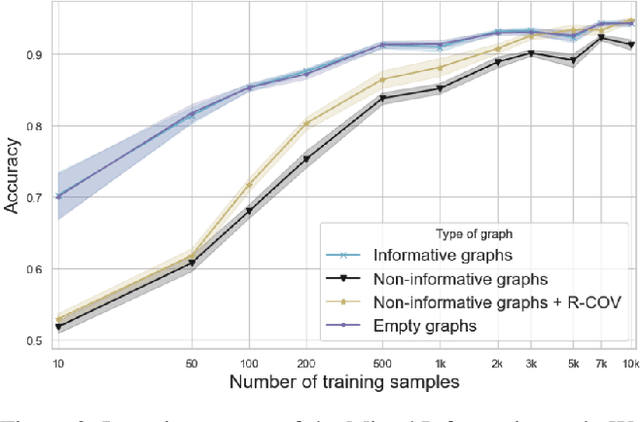
Predictions over graphs play a crucial role in various domains, including social networks, molecular biology, medicine, and more. Graph Neural Networks (GNNs) have emerged as the dominant approach for learning on graph data. Instances of graph labeling problems consist of the graph-structure (i.e., the adjacency matrix), along with node-specific feature vectors. In some cases, this graph-structure is non-informative for the predictive task. For instance, molecular properties such as molar mass depend solely on the constituent atoms (node features), and not on the molecular structure. While GNNs have the ability to ignore the graph-structure in such cases, it is not clear that they will. In this work, we show that GNNs actually tend to overfit the graph-structure in the sense that they use it even when a better solution can be obtained by ignoring it. We examine this phenomenon with respect to different graph distributions and find that regular graphs are more robust to this overfitting. We then provide a theoretical explanation for this phenomenon, via analyzing the implicit bias of gradient-descent-based learning of GNNs in this setting. Finally, based on our empirical and theoretical findings, we propose a graph-editing method to mitigate the tendency of GNNs to overfit graph-structures that should be ignored. We show that this method indeed improves the accuracy of GNNs across multiple benchmarks.