 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBayesian Reasoning Enabled by Spin-Orbit Torque Magnetic Tunnel Junctions
Apr 11, 2025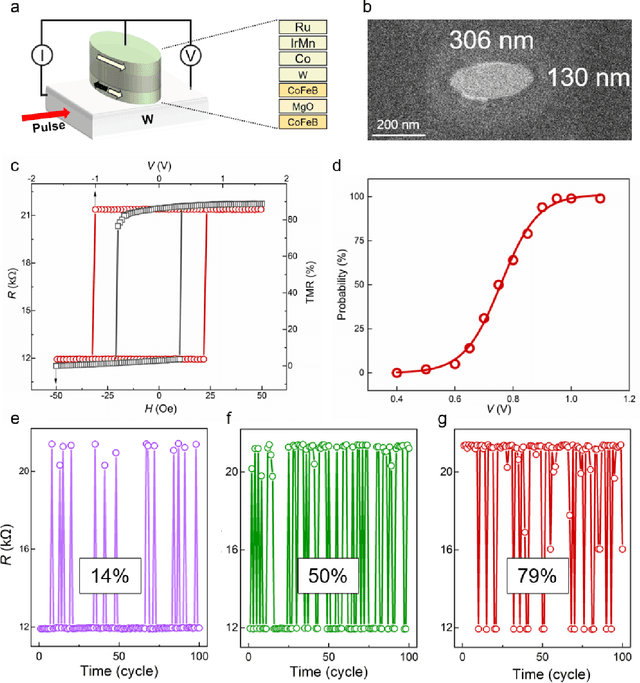
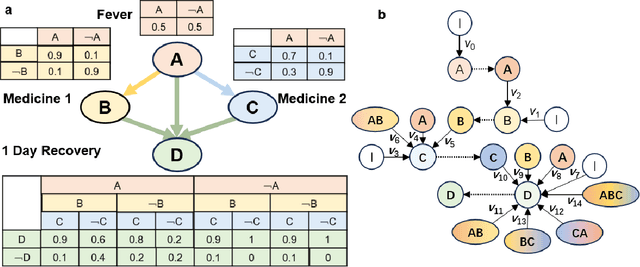
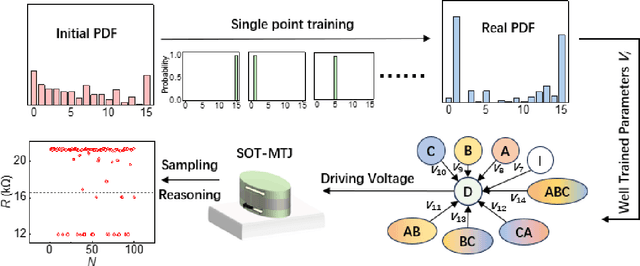
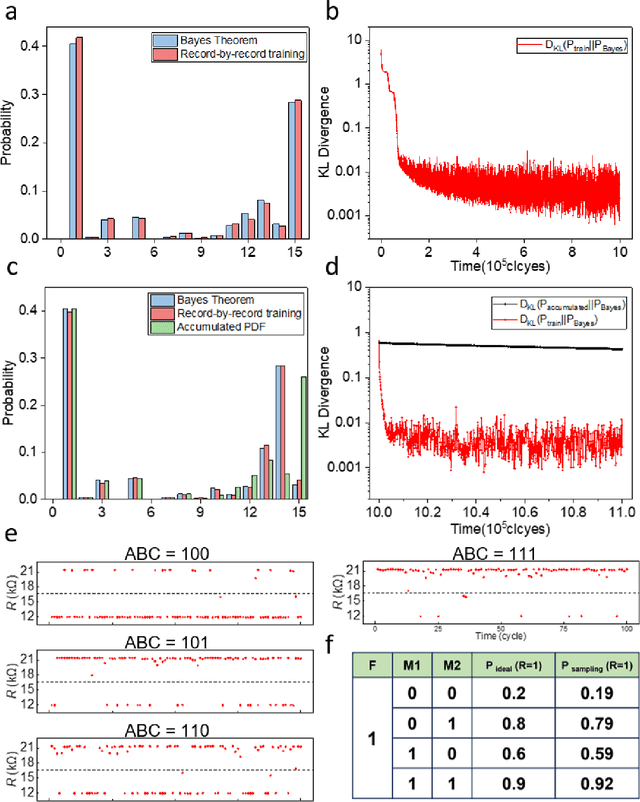
Bayesian networks play an increasingly important role in data mining, inference, and reasoning with the rapid development of artificial intelligence. In this paper, we present proof-of-concept experiments demonstrating the use of spin-orbit torque magnetic tunnel junctions (SOT-MTJs) in Bayesian network reasoning. Not only can the target probability distribution function (PDF) of a Bayesian network be precisely formulated by a conditional probability table as usual but also quantitatively parameterized by a probabilistic forward propagating neuron network. Moreover, the parameters of the network can also approach the optimum through a simple point-by point training algorithm, by leveraging which we do not need to memorize all historical data nor statistically summarize conditional probabilities behind them, significantly improving storage efficiency and economizing data pretreatment. Furthermore, we developed a simple medical diagnostic system using the SOT-MTJ as a random number generator and sampler, showcasing the application of SOT-MTJ-based Bayesian reasoning. This SOT-MTJ-based Bayesian reasoning shows great promise in the field of artificial probabilistic neural network, broadening the scope of spintronic device applications and providing an efficient and low-storage solution for complex reasoning tasks.
Latent Diffusion Model for Medical Image Standardization and Enhancement
Oct 08, 2023



Computed tomography (CT) serves as an effective tool for lung cancer screening, diagnosis, treatment, and prognosis, providing a rich source of features to quantify temporal and spatial tumor changes. Nonetheless, the diversity of CT scanners and customized acquisition protocols can introduce significant inconsistencies in texture features, even when assessing the same patient. This variability poses a fundamental challenge for subsequent research that relies on consistent image features. Existing CT image standardization models predominantly utilize GAN-based supervised or semi-supervised learning, but their performance remains limited. We present DiffusionCT, an innovative score-based DDPM model that operates in the latent space to transform disparate non-standard distributions into a standardized form. The architecture comprises a U-Net-based encoder-decoder, augmented by a DDPM model integrated at the bottleneck position. First, the encoder-decoder is trained independently, without embedding DDPM, to capture the latent representation of the input data. Second, the latent DDPM model is trained while keeping the encoder-decoder parameters fixed. Finally, the decoder uses the transformed latent representation to generate a standardized CT image, providing a more consistent basis for downstream analysis. Empirical tests on patient CT images indicate notable improvements in image standardization using DiffusionCT. Additionally, the model significantly reduces image noise in SPAD images, further validating the effectiveness of DiffusionCT for advanced imaging tasks.
Efficient Global Multi-object Tracking Under Minimum-cost Circulation Framework
Nov 02, 2019
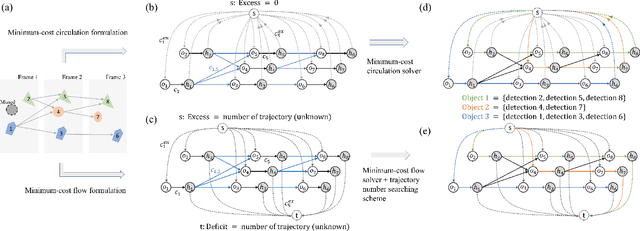

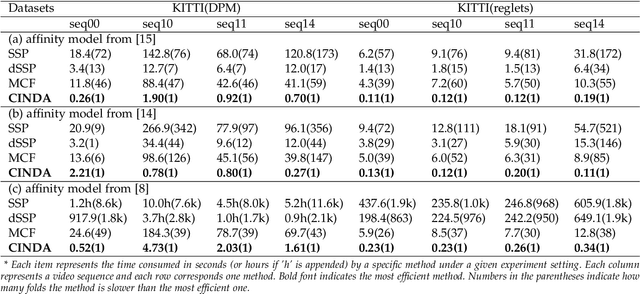
We developed a minimum-cost circulation framework for solving the global data association problem, which plays a key role in the tracking-by-detection paradigm of multi-object tracking. The global data association problem was extensively studied under the minimum-cost flow framework, which is theoretically attractive as being flexible and globally solvable. However, the high computational burden has been a long-standing obstacle to its wide adoption in practice. While enjoying the same theoretical advantages and maintaining the same optimal solution as the minimum-cost flow framework, our new framework has a better theoretical complexity bound and leads to orders of practical efficiency improvement. This new framework is motivated by the observation that minimum-cost flow only partially models the data association problem and must be accompanied by an additional and time-consuming searching scheme to determine the optimal object number. By employing a minimum-cost circulation framework, we eliminate the searching step and naturally integrate the number of objects into the optimization problem. By exploring the special property of the associated graph, that is, an overwhelming majority of the vertices are with unit capacity, we designed an implementation of the framework and proved it has the best theoretical complexity so far for the global data association problem. We evaluated our method with 40 experiments on five MOT benchmark datasets. Our method was always the most efficient and averagely 53 to 1,192 times faster than the three state-of-the-art methods. When our method served as a sub-module for global data association methods using higher-order constraints, similar efficiency improvement was attained. We further illustrated through several case studies how the improved computational efficiency enables more sophisticated tracking models and yields better tracking accuracy.
A feasible roadmap for unsupervised deconvolution of two-source mixed gene expressions
Oct 25, 2013
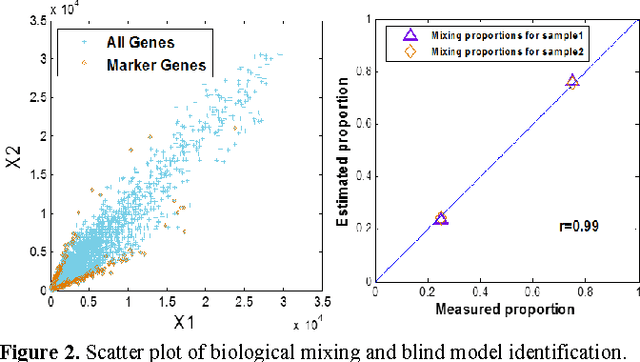


Tissue heterogeneity is a major confounding factor in studying individual populations that cannot be resolved directly by global profiling. Experimental solutions to mitigate tissue heterogeneity are expensive, time consuming, inapplicable to existing data, and may alter the original gene expression patterns. Here we ask whether it is possible to deconvolute two-source mixed expressions (estimating both proportions and cell-specific profiles) from two or more heterogeneous samples without requiring any prior knowledge. Supported by a well-grounded mathematical framework, we argue that both constituent proportions and cell-specific expressions can be estimated in a completely unsupervised mode when cell-specific marker genes exist, which do not have to be known a priori, for each of constituent cell types. We demonstrate the performance of unsupervised deconvolution on both simulation and real gene expression data, together with perspective discussions.