 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConvex Analysis of Mixtures for Separating Non-negative Well-grounded Sources
Dec 10, 2015
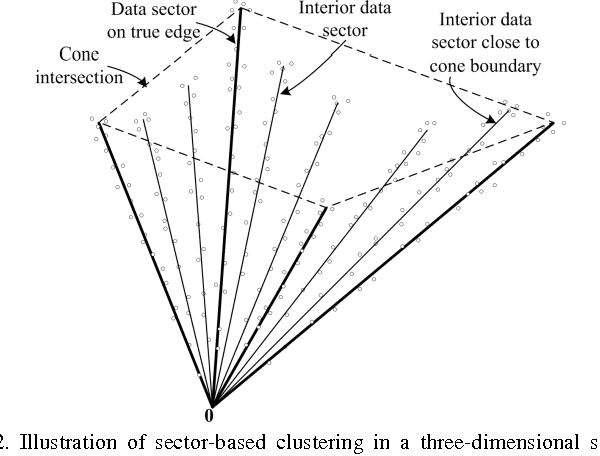


Blind Source Separation (BSS) has proven to be a powerful tool for the analysis of composite patterns in engineering and science. We introduce Convex Analysis of Mixtures (CAM) for separating non-negative well-grounded sources, which learns the mixing matrix by identifying the lateral edges of the convex data scatter plot. We prove a sufficient and necessary condition for identifying the mixing matrix through edge detection, which also serves as the foundation for CAM to be applied not only to the exact-determined and over-determined cases, but also to the under-determined case. We show the optimality of the edge detection strategy, even for cases where source well-groundedness is not strictly satisfied. The CAM algorithm integrates plug-in noise filtering using sector-based clustering, an efficient geometric convex analysis scheme, and stability-based model order selection. We demonstrate the principle of CAM on simulated data and numerically mixed natural images. The superior performance of CAM against a panel of benchmark BSS techniques is demonstrated on numerically mixed gene expression data. We then apply CAM to dissect dynamic contrast-enhanced magnetic resonance imaging data taken from breast tumors and time-course microarray gene expression data derived from in-vivo muscle regeneration in mice, both producing biologically plausible decomposition results.
Unsupervised deconvolution of dynamic imaging reveals intratumor vascular heterogeneity
Sep 04, 2014Intratumor heterogeneity is often manifested by vascular compartments with distinct pharmacokinetics that cannot be resolved directly by in vivo dynamic imaging. We developed tissue-specific compartment modeling (TSCM), an unsupervised computational method of deconvolving dynamic imaging series from heterogeneous tumors that can improve vascular phenotyping in many biological contexts. Applying TSCM to dynamic contrast-enhanced MRI of breast cancers revealed characteristic intratumor vascular heterogeneity and therapeutic responses that were otherwise undetectable.
A feasible roadmap for unsupervised deconvolution of two-source mixed gene expressions
Oct 25, 2013
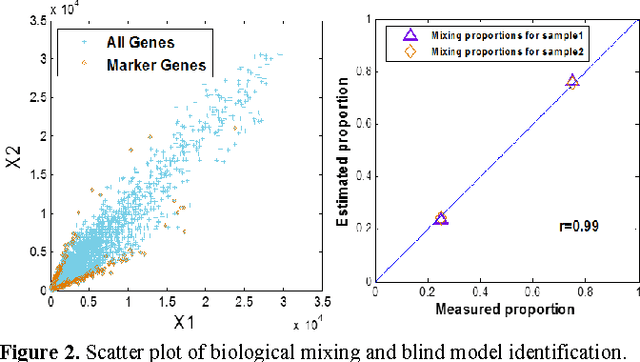


Tissue heterogeneity is a major confounding factor in studying individual populations that cannot be resolved directly by global profiling. Experimental solutions to mitigate tissue heterogeneity are expensive, time consuming, inapplicable to existing data, and may alter the original gene expression patterns. Here we ask whether it is possible to deconvolute two-source mixed expressions (estimating both proportions and cell-specific profiles) from two or more heterogeneous samples without requiring any prior knowledge. Supported by a well-grounded mathematical framework, we argue that both constituent proportions and cell-specific expressions can be estimated in a completely unsupervised mode when cell-specific marker genes exist, which do not have to be known a priori, for each of constituent cell types. We demonstrate the performance of unsupervised deconvolution on both simulation and real gene expression data, together with perspective discussions.