 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProof of Concept: Multi-Target Wildfire Risk Prediction and Large Language Model Synthesis
Jan 16, 2026Current state-of-the-art approaches to wildfire risk assessment often overlook operational needs, limiting their practical value for first responders and firefighting services. Effective wildfire management requires a multi-target analysis that captures the diverse dimensions of wildfire risk, including meteorological danger, ignition activity, intervention complexity, and resource mobilization, rather than relying on a single predictive indicator. In this proof of concept, we propose the development of a hybrid framework that combines predictive models for each risk dimension with large language models (LLMs) to synthesize heterogeneous outputs into structured, actionable reports.
Extreme-value forest fire prediction A study of the Loss Function in an Ordinality Scheme
Jan 08, 2026Wildfires are highly imbalanced natural hazards in both space and severity, making the prediction of extreme events particularly challenging. In this work, we introduce the first ordinal classification framework for forecasting wildfire severity levels directly aligned with operational decision-making in France. Our study investigates the influence of loss-function design on the ability of neural models to predict rare yet critical high-severity fire occurrences. We compare standard cross-entropy with several ordinal-aware objectives, including the proposed probabilistic TDeGPD loss derived from a truncated discrete exponentiated Generalized Pareto Distribution. Through extensive benchmarking over multiple architectures and real operational data, we show that ordinal supervision substantially improves model performance over conventional approaches. In particular, the Weighted Kappa Loss (WKLoss) achieves the best overall results, with more than +0.1 IoU (Intersection Over Union) gain on the most extreme severity classes while maintaining competitive calibration quality. However, performance remains limited for the rarest events due to their extremely low representation in the dataset. These findings highlight the importance of integrating both severity ordering, data imbalance considerations, and seasonality risk into wildfire forecasting systems. Future work will focus on incorporating seasonal dynamics and uncertainty information into training to further improve the reliability of extreme-event prediction.
De-Identification of French Unstructured Clinical Notes for Machine Learning Tasks
Sep 16, 2022
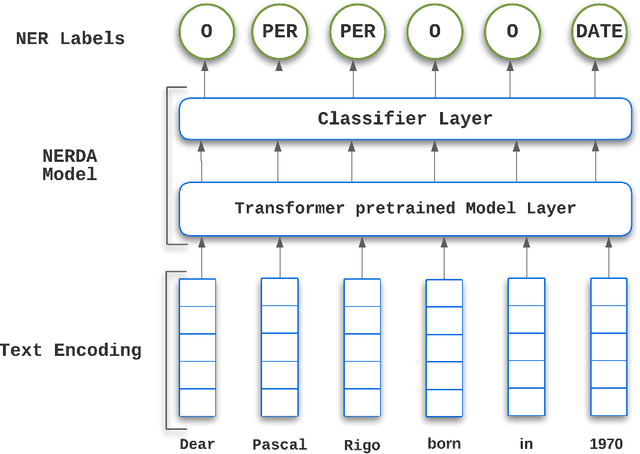
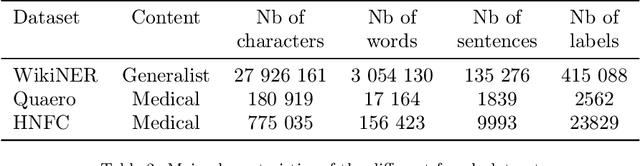
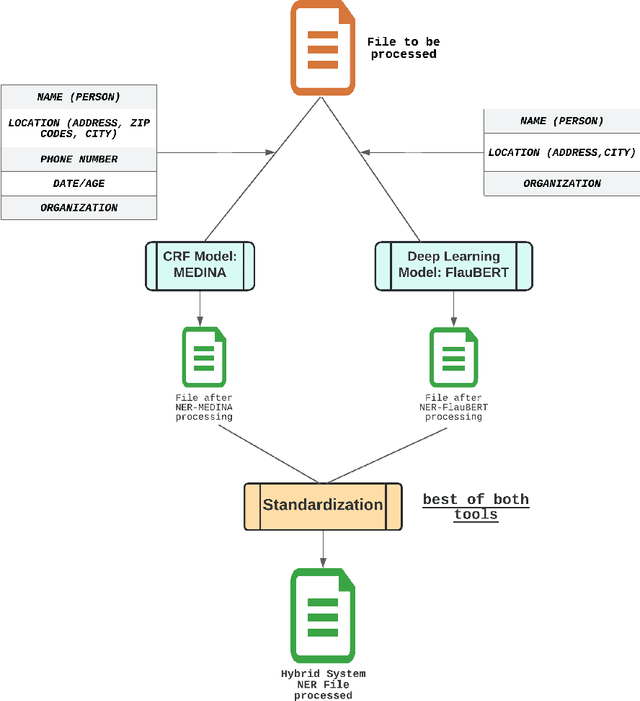
Unstructured textual data are at the heart of health systems: liaison letters between doctors, operating reports, coding of procedures according to the ICD-10 standard, etc. The details included in these documents make it possible to get to know the patient better, to better manage him or her, to better study the pathologies, to accurately remunerate the associated medical acts\ldots All this seems to be (at least partially) within reach of today by artificial intelligence techniques. However, for obvious reasons of privacy protection, the designers of these AIs do not have the legal right to access these documents as long as they contain identifying data. De-identifying these documents, i.e. detecting and deleting all identifying information present in them, is a legally necessary step for sharing this data between two complementary worlds. Over the last decade, several proposals have been made to de-identify documents, mainly in English. While the detection scores are often high, the substitution methods are often not very robust to attack. In French, very few methods are based on arbitrary detection and/or substitution rules. In this paper, we propose a new comprehensive de-identification method dedicated to French-language medical documents. Both the approach for the detection of identifying elements (based on deep learning) and their substitution (based on differential privacy) are based on the most proven existing approaches. The result is an approach that effectively protects the privacy of the patients at the heart of these medical documents. The whole approach has been evaluated on a French language medical dataset of a French public hospital and the results are very encouraging.
Average performance analysis of the stochastic gradient method for online PCA
Apr 03, 2018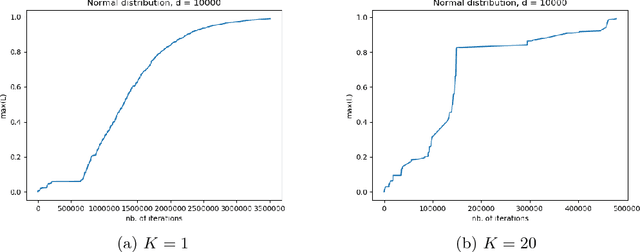
This paper studies the complexity of the stochastic gradient algorithm for PCA when the data are observed in a streaming setting. We also propose an online approach for selecting the learning rate. Simulation experiments confirm the practical relevance of the plain stochastic gradient approach and that drastic improvements can be achieved by learning the learning rate.
Well-supported phylogenies using largest subsets of core-genes by discrete particle swarm optimization
Jun 25, 2017

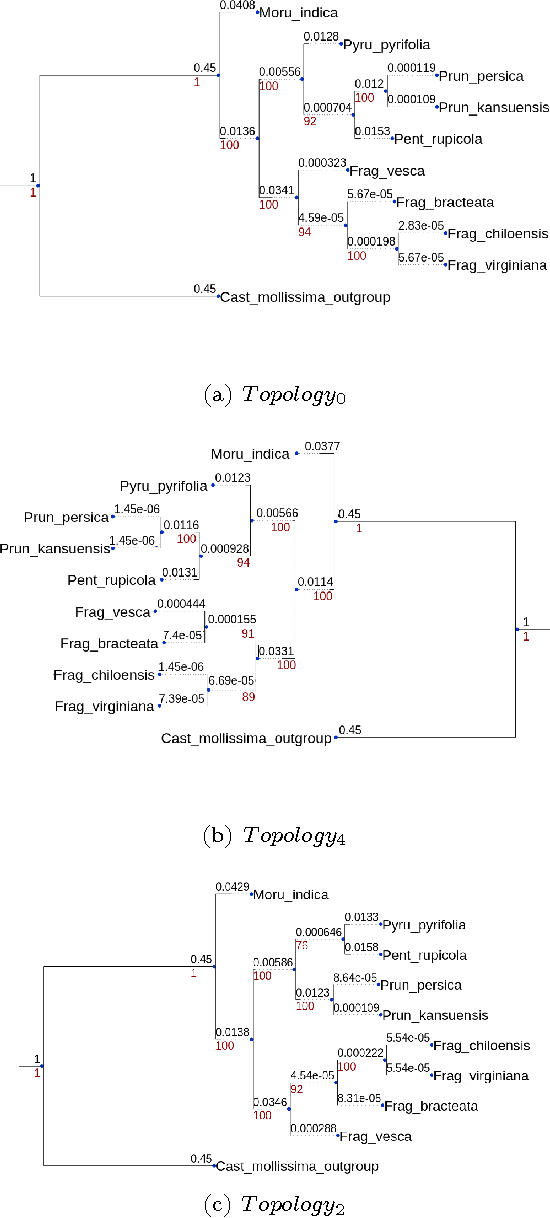
The number of complete chloroplastic genomes increases day after day, making it possible to rethink plants phylogeny at the biomolecular era. Given a set of close plants sharing in the order of one hundred of core chloroplastic genes, this article focuses on how to extract the largest subset of sequences in order to obtain the most supported species tree. Due to computational complexity, a discrete and distributed Particle Swarm Optimization (DPSO) is proposed. It is finally applied to the core genes of Rosales order.
Random Forests for Industrial Device Functioning Diagnostics Using Wireless Sensor Networks
Jun 25, 2017


In this paper, random forests are proposed for operating devices diagnostics in the presence of a variable number of features. In various contexts, like large or difficult-to-access monitored areas, wired sensor networks providing features to achieve diagnostics are either very costly to use or totally impossible to spread out. Using a wireless sensor network can solve this problem, but this latter is more subjected to flaws. Furthermore, the networks' topology often changes, leading to a variability in quality of coverage in the targeted area. Diagnostics at the sink level must take into consideration that both the number and the quality of the provided features are not constant, and that some politics like scheduling or data aggregation may be developed across the network. The aim of this article is ($1$) to show that random forests are relevant in this context, due to their flexibility and robustness, and ($2$) to provide first examples of use of this method for diagnostics based on data provided by a wireless sensor network.
Finding optimal finite biological sequences over finite alphabets: the OptiFin toolbox
Jun 25, 2017

In this paper, we present a toolbox for a specific optimization problem that frequently arises in bioinformatics or genomics. In this specific optimisation problem, the state space is a set of words of specified length over a finite alphabet. To each word is associated a score. The overall objective is to find the words which have the lowest possible score. This type of general optimization problem is encountered in e.g 3D conformation optimisation for protein structure prediction, or largest core genes subset discovery based on best supported phylogenetic tree for a set of species. In order to solve this problem, we propose a toolbox that can be easily launched using MPI and embeds 3 well-known metaheuristics. The toolbox is fully parametrized and well documented. It has been specifically designed to be easy modified and possibly improved by the user depending on the application, and does not require to be a computer scientist. We show that the toolbox performs very well on two difficult practical problems.
Efficient and accurate monitoring of the depth information in a Wireless Multimedia Sensor Network based surveillance
Jun 25, 2017

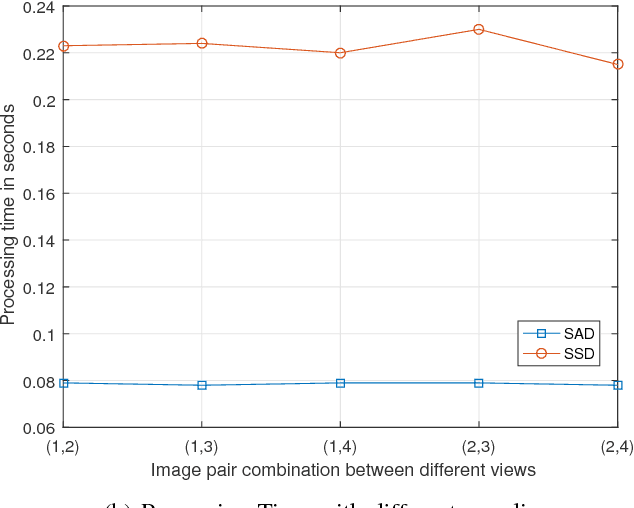
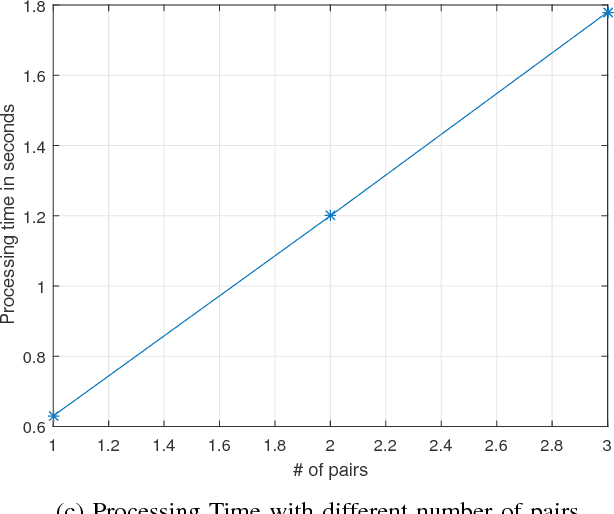
Wireless Multimedia Sensor Network (WMSN) is a promising technology capturing rich multimedia data like audio and video, which can be useful to monitor an environment under surveillance. However, many scenarios in real time monitoring requires 3D depth information. In this research work, we propose to use the disparity map that is computed from two or multiple images, in order to monitor the depth information in an object or event under surveillance using WMSN. Our system is based on distributed wireless sensors allowing us to notably reduce the computational time needed for 3D depth reconstruction, thus permitting the success of real time solutions. Each pair of sensors will capture images for a targeted place/object and will operate a Stereo Matching in order to create a Disparity Map. Disparity maps will give us the ability to decrease traffic on the bandwidth, because they are of low size. This will increase WMSN lifetime. Any event can be detected after computing the depth value for the target object in the scene, and also 3D scene reconstruction can be achieved with a disparity map and some reference(s) image(s) taken by the node(s).
Binary Particle Swarm Optimization versus Hybrid Genetic Algorithm for Inferring Well Supported Phylogenetic Trees
Aug 31, 2016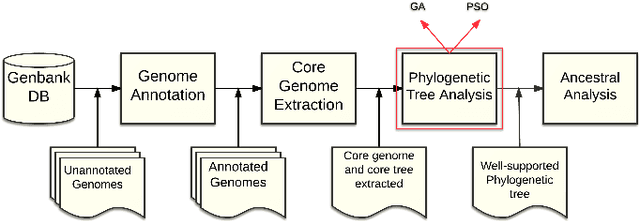
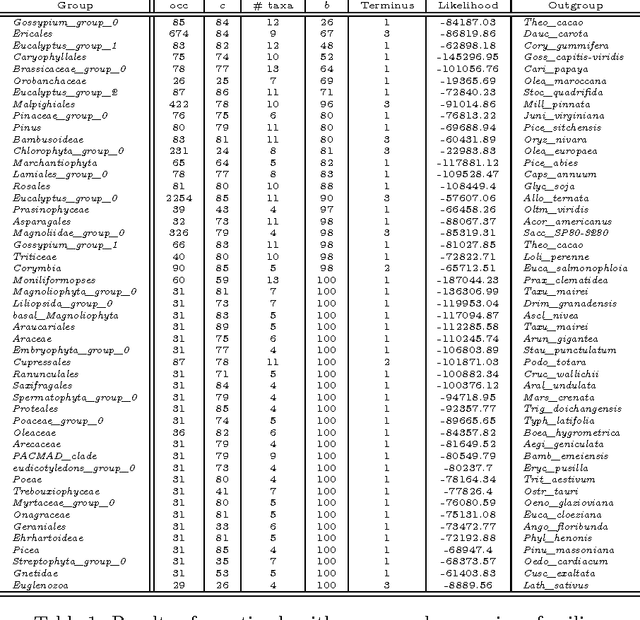

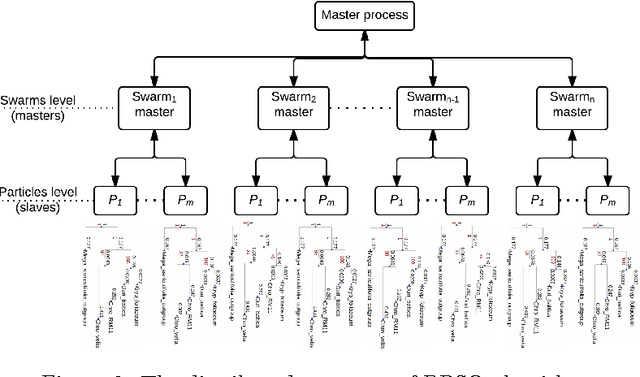
The amount of completely sequenced chloroplast genomes increases rapidly every day, leading to the possibility to build large-scale phylogenetic trees of plant species. Considering a subset of close plant species defined according to their chloroplasts, the phylogenetic tree that can be inferred by their core genes is not necessarily well supported, due to the possible occurrence of problematic genes (i.e., homoplasy, incomplete lineage sorting, horizontal gene transfers, etc.) which may blur the phylogenetic signal. However, a trustworthy phylogenetic tree can still be obtained provided such a number of blurring genes is reduced. The problem is thus to determine the largest subset of core genes that produces the best-supported tree. To discard problematic genes and due to the overwhelming number of possible combinations, this article focuses on how to extract the largest subset of sequences in order to obtain the most supported species tree. Due to computational complexity, a distributed Binary Particle Swarm Optimization (BPSO) is proposed in sequential and distributed fashions. Obtained results from both versions of the BPSO are compared with those computed using an hybrid approach embedding both genetic algorithms and statistical tests. The proposal has been applied to different cases of plant families, leading to encouraging results for these families.
Neural Networks and Chaos: Construction, Evaluation of Chaotic Networks, and Prediction of Chaos with Multilayer Feedforward Networks
Aug 21, 2016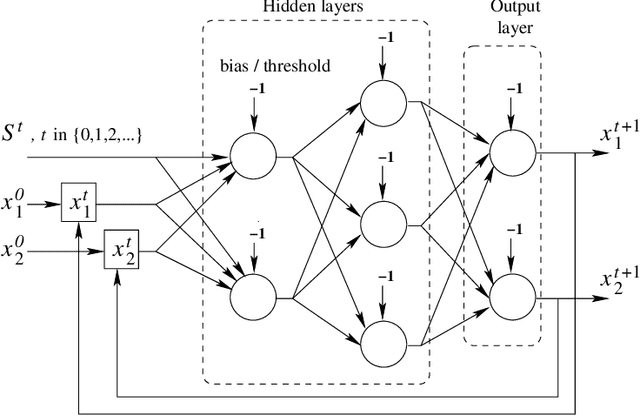
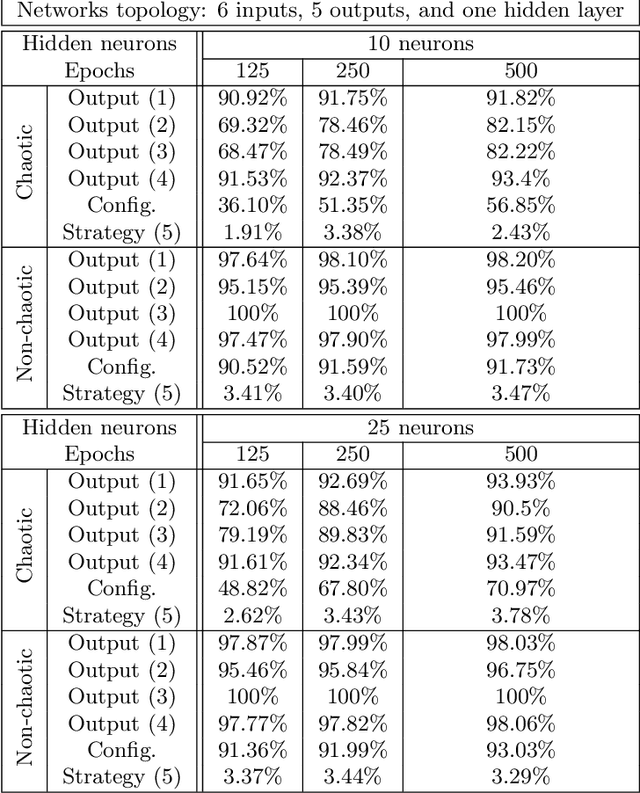
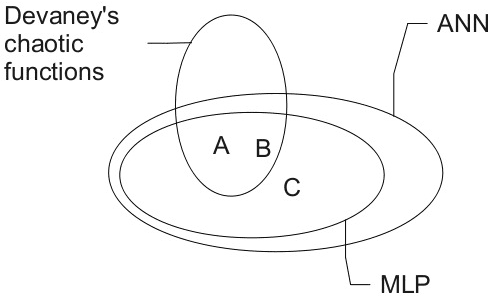

Many research works deal with chaotic neural networks for various fields of application. Unfortunately, up to now these networks are usually claimed to be chaotic without any mathematical proof. The purpose of this paper is to establish, based on a rigorous theoretical framework, an equivalence between chaotic iterations according to Devaney and a particular class of neural networks. On the one hand we show how to build such a network, on the other hand we provide a method to check if a neural network is a chaotic one. Finally, the ability of classical feedforward multilayer perceptrons to learn sets of data obtained from a dynamical system is regarded. Various Boolean functions are iterated on finite states. Iterations of some of them are proven to be chaotic as it is defined by Devaney. In that context, important differences occur in the training process, establishing with various neural networks that chaotic behaviors are far more difficult to learn.