 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHybrid Genetic Algorithm and Lasso Test Approach for Inferring Well Supported Phylogenetic Trees based on Subsets of Chloroplastic Core Genes
Apr 20, 2015
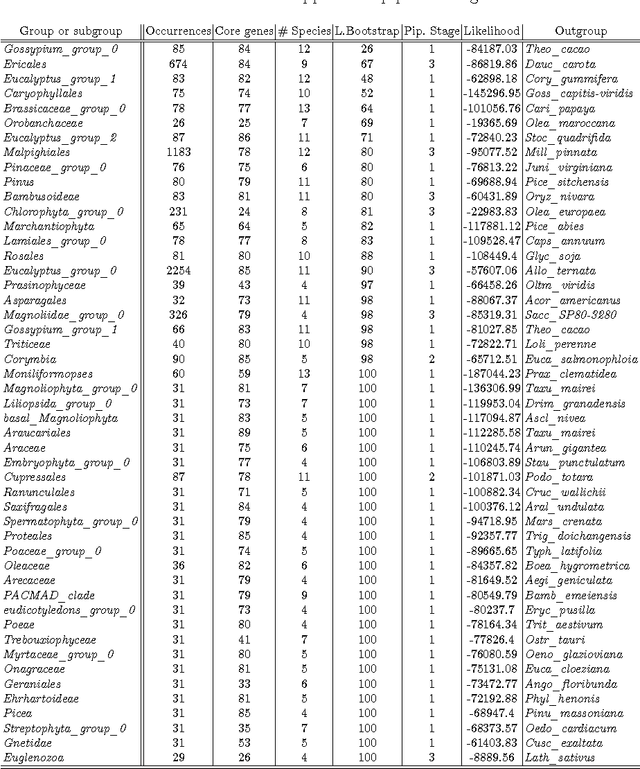
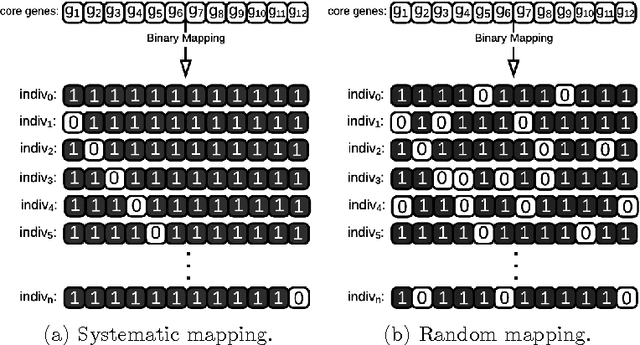
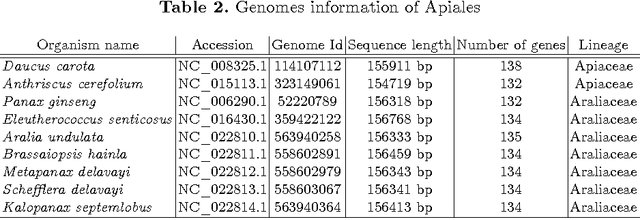
The amount of completely sequenced chloroplast genomes increases rapidly every day, leading to the possibility to build large scale phylogenetic trees of plant species. Considering a subset of close plant species defined according to their chloroplasts, the phylogenetic tree that can be inferred by their core genes is not necessarily well supported, due to the possible occurrence of "problematic" genes (i.e., homoplasy, incomplete lineage sorting, horizontal gene transfers, etc.) which may blur phylogenetic signal. However, a trustworthy phylogenetic tree can still be obtained if the number of problematic genes is low, the problem being to determine the largest subset of core genes that produces the best supported tree. To discard problematic genes and due to the overwhelming number of possible combinations, we propose an hybrid approach that embeds both genetic algorithms and statistical tests. Given a set of organisms, the result is a pipeline of many stages for the production of well supported phylogenetic trees. The proposal has been applied to different cases of plant families, leading to encouraging results for these families.