 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomatic ICD-10 Code Association: A Challenging Task on French Clinical Texts
Apr 06, 2023



Automatically associating ICD codes with electronic health data is a well-known NLP task in medical research. NLP has evolved significantly in recent years with the emergence of pre-trained language models based on Transformers architecture, mainly in the English language. This paper adapts these models to automatically associate the ICD codes. Several neural network architectures have been experimented with to address the challenges of dealing with a large set of both input tokens and labels to be guessed. In this paper, we propose a model that combines the latest advances in NLP and multi-label classification for ICD-10 code association. Fair experiments on a Clinical dataset in the French language show that our approach increases the $F_1$-score metric by more than 55\% compared to state-of-the-art results.
An Easy-to-use and Robust Approach for the Differentially Private De-Identification of Clinical Textual Documents
Nov 02, 2022Unstructured textual data is at the heart of healthcare systems. For obvious privacy reasons, these documents are not accessible to researchers as long as they contain personally identifiable information. One way to share this data while respecting the legislative framework (notably GDPR or HIPAA) is, within the medical structures, to de-identify it, i.e. to detect the personal information of a person through a Named Entity Recognition (NER) system and then replacing it to make it very difficult to associate the document with the person. The challenge is having reliable NER and substitution tools without compromising confidentiality and consistency in the document. Most of the conducted research focuses on English medical documents with coarse substitutions by not benefiting from advances in privacy. This paper shows how an efficient and differentially private de-identification approach can be achieved by strengthening the less robust de-identification method and by adapting state-of-the-art differentially private mechanisms for substitution purposes. The result is an approach for de-identifying clinical documents in French language, but also generalizable to other languages and whose robustness is mathematically proven.
De-Identification of French Unstructured Clinical Notes for Machine Learning Tasks
Sep 16, 2022
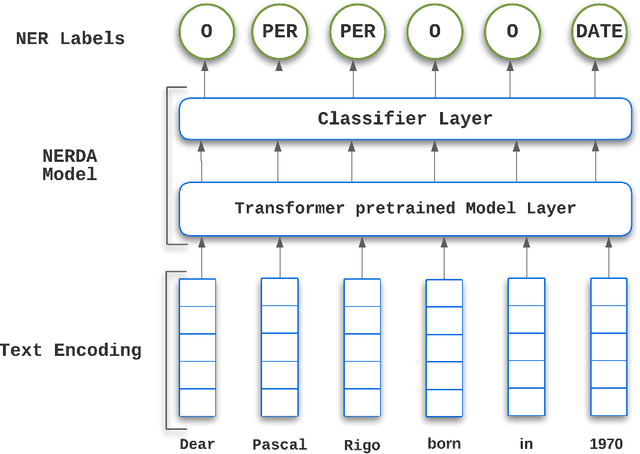
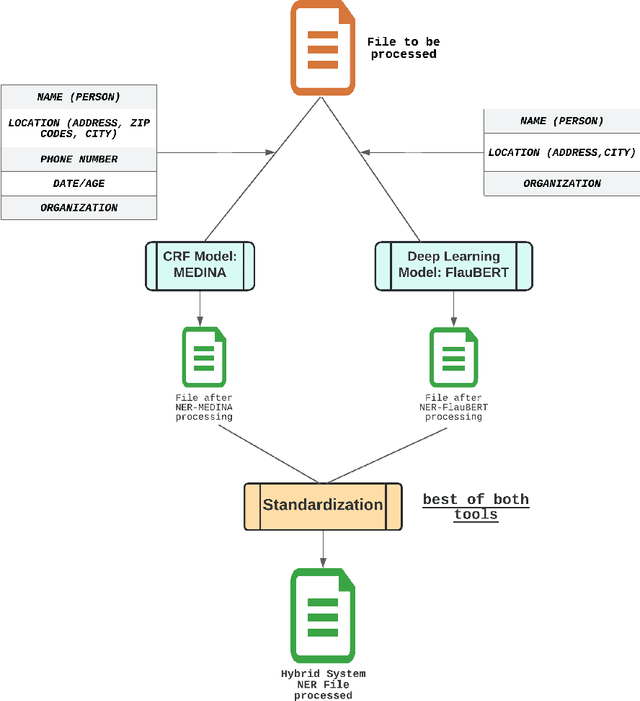
Unstructured textual data are at the heart of health systems: liaison letters between doctors, operating reports, coding of procedures according to the ICD-10 standard, etc. The details included in these documents make it possible to get to know the patient better, to better manage him or her, to better study the pathologies, to accurately remunerate the associated medical acts\ldots All this seems to be (at least partially) within reach of today by artificial intelligence techniques. However, for obvious reasons of privacy protection, the designers of these AIs do not have the legal right to access these documents as long as they contain identifying data. De-identifying these documents, i.e. detecting and deleting all identifying information present in them, is a legally necessary step for sharing this data between two complementary worlds. Over the last decade, several proposals have been made to de-identify documents, mainly in English. While the detection scores are often high, the substitution methods are often not very robust to attack. In French, very few methods are based on arbitrary detection and/or substitution rules. In this paper, we propose a new comprehensive de-identification method dedicated to French-language medical documents. Both the approach for the detection of identifying elements (based on deep learning) and their substitution (based on differential privacy) are based on the most proven existing approaches. The result is an approach that effectively protects the privacy of the patients at the heart of these medical documents. The whole approach has been evaluated on a French language medical dataset of a French public hospital and the results are very encouraging.
Differentially Private Multivariate Time Series Forecasting of Aggregated Human Mobility With Deep Learning: Input or Gradient Perturbation?
May 01, 2022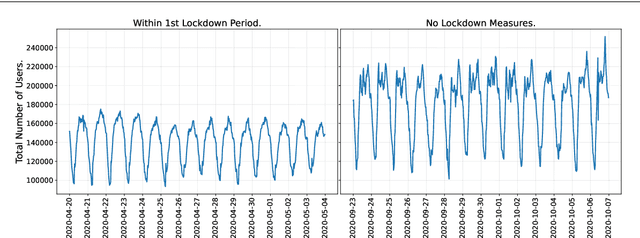
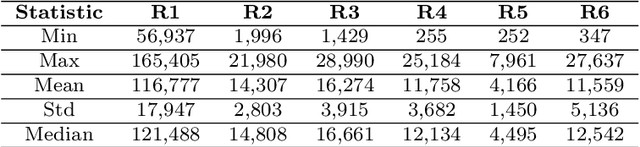
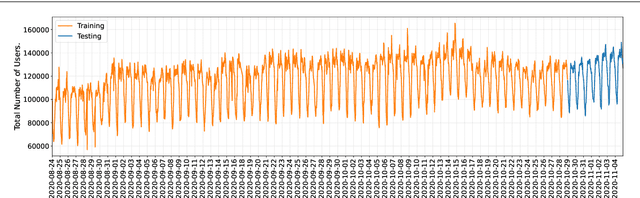
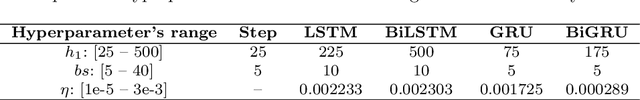
This paper investigates the problem of forecasting multivariate aggregated human mobility while preserving the privacy of the individuals concerned. Differential privacy, a state-of-the-art formal notion, has been used as the privacy guarantee in two different and independent steps when training deep learning models. On one hand, we considered \textit{gradient perturbation}, which uses the differentially private stochastic gradient descent algorithm to guarantee the privacy of each time series sample in the learning stage. On the other hand, we considered \textit{input perturbation}, which adds differential privacy guarantees in each sample of the series before applying any learning. We compared four state-of-the-art recurrent neural networks: Long Short-Term Memory, Gated Recurrent Unit, and their Bidirectional architectures, i.e., Bidirectional-LSTM and Bidirectional-GRU. Extensive experiments were conducted with a real-world multivariate mobility dataset, which we published openly along with this paper. As shown in the results, differentially private deep learning models trained under gradient or input perturbation achieve nearly the same performance as non-private deep learning models, with loss in performance varying between $0.57\%$ to $2.8\%$. The contribution of this paper is significant for those involved in urban planning and decision-making, providing a solution to the human mobility multivariate forecast problem through differentially private deep learning models.
Well-supported phylogenies using largest subsets of core-genes by discrete particle swarm optimization
Jun 25, 2017

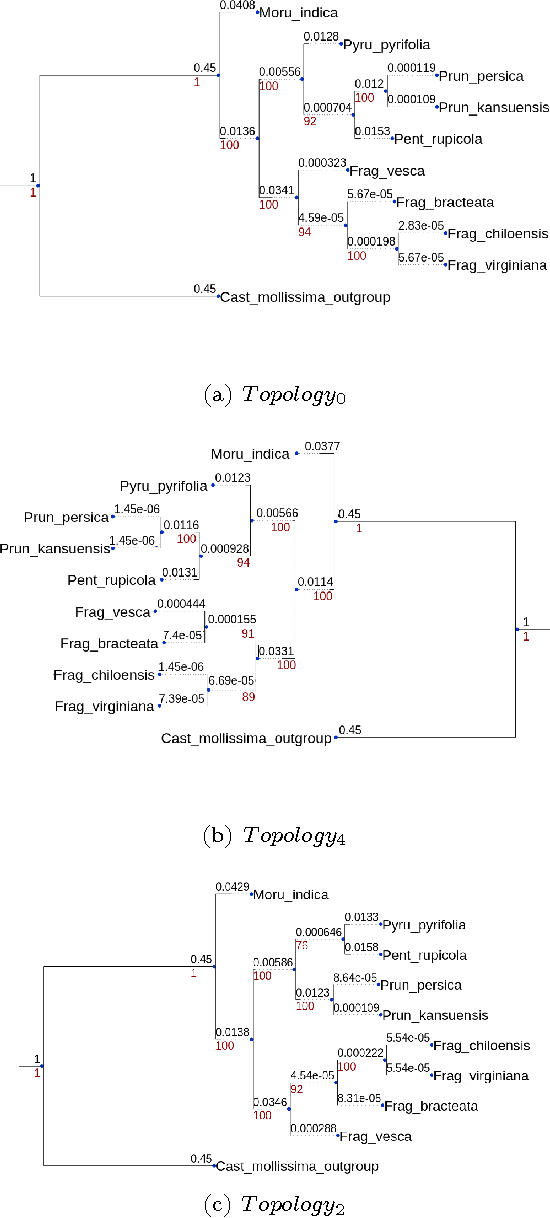
The number of complete chloroplastic genomes increases day after day, making it possible to rethink plants phylogeny at the biomolecular era. Given a set of close plants sharing in the order of one hundred of core chloroplastic genes, this article focuses on how to extract the largest subset of sequences in order to obtain the most supported species tree. Due to computational complexity, a discrete and distributed Particle Swarm Optimization (DPSO) is proposed. It is finally applied to the core genes of Rosales order.
Binary Particle Swarm Optimization versus Hybrid Genetic Algorithm for Inferring Well Supported Phylogenetic Trees
Aug 31, 2016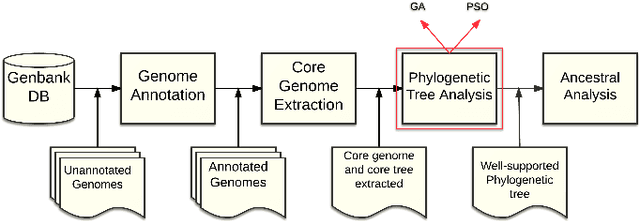
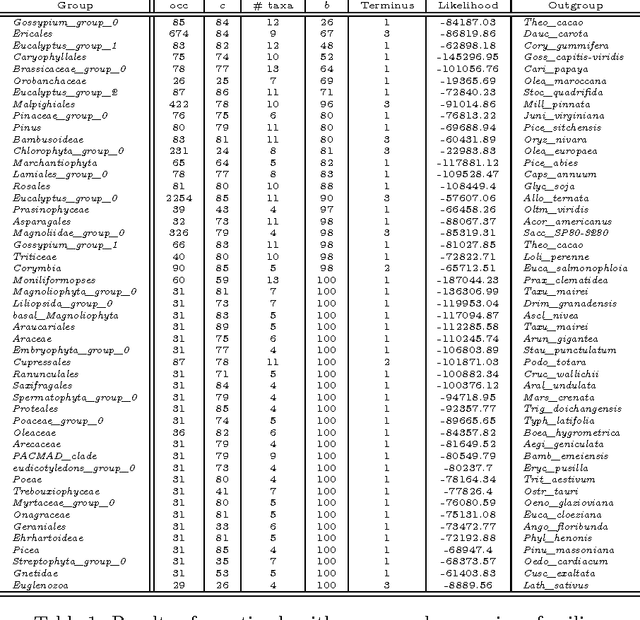

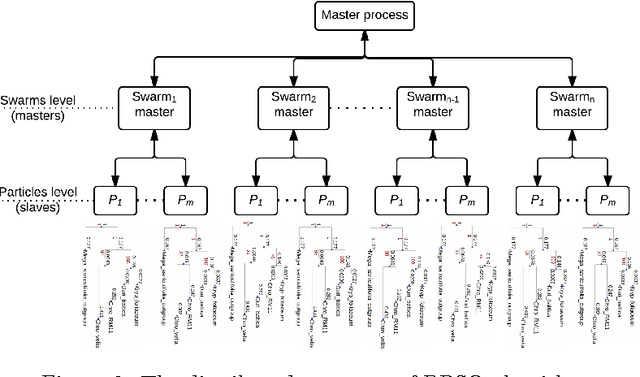
The amount of completely sequenced chloroplast genomes increases rapidly every day, leading to the possibility to build large-scale phylogenetic trees of plant species. Considering a subset of close plant species defined according to their chloroplasts, the phylogenetic tree that can be inferred by their core genes is not necessarily well supported, due to the possible occurrence of problematic genes (i.e., homoplasy, incomplete lineage sorting, horizontal gene transfers, etc.) which may blur the phylogenetic signal. However, a trustworthy phylogenetic tree can still be obtained provided such a number of blurring genes is reduced. The problem is thus to determine the largest subset of core genes that produces the best-supported tree. To discard problematic genes and due to the overwhelming number of possible combinations, this article focuses on how to extract the largest subset of sequences in order to obtain the most supported species tree. Due to computational complexity, a distributed Binary Particle Swarm Optimization (BPSO) is proposed in sequential and distributed fashions. Obtained results from both versions of the BPSO are compared with those computed using an hybrid approach embedding both genetic algorithms and statistical tests. The proposal has been applied to different cases of plant families, leading to encouraging results for these families.
Neural Networks and Chaos: Construction, Evaluation of Chaotic Networks, and Prediction of Chaos with Multilayer Feedforward Networks
Aug 21, 2016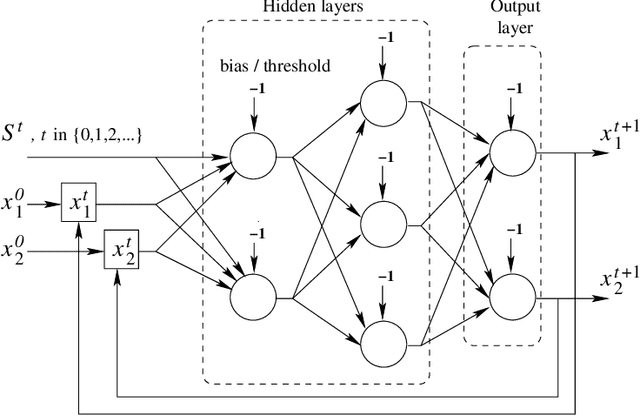
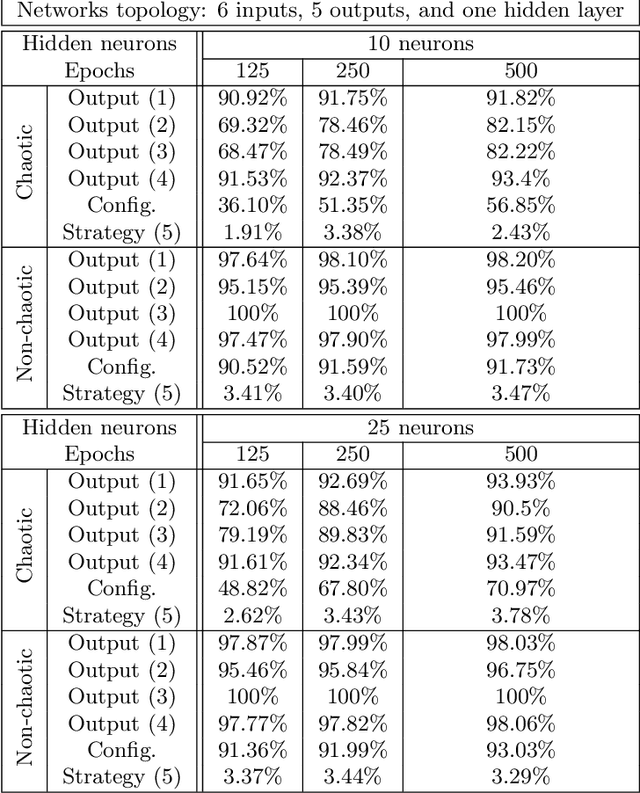
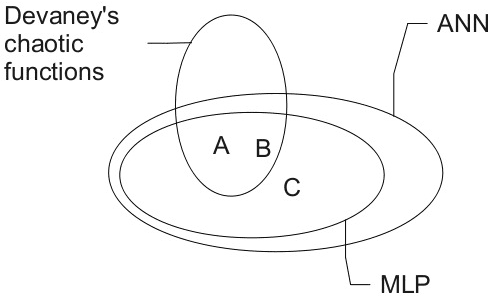

Many research works deal with chaotic neural networks for various fields of application. Unfortunately, up to now these networks are usually claimed to be chaotic without any mathematical proof. The purpose of this paper is to establish, based on a rigorous theoretical framework, an equivalence between chaotic iterations according to Devaney and a particular class of neural networks. On the one hand we show how to build such a network, on the other hand we provide a method to check if a neural network is a chaotic one. Finally, the ability of classical feedforward multilayer perceptrons to learn sets of data obtained from a dynamical system is regarded. Various Boolean functions are iterated on finite states. Iterations of some of them are proven to be chaotic as it is defined by Devaney. In that context, important differences occur in the training process, establishing with various neural networks that chaotic behaviors are far more difficult to learn.
Hybrid Genetic Algorithm and Lasso Test Approach for Inferring Well Supported Phylogenetic Trees based on Subsets of Chloroplastic Core Genes
Apr 20, 2015
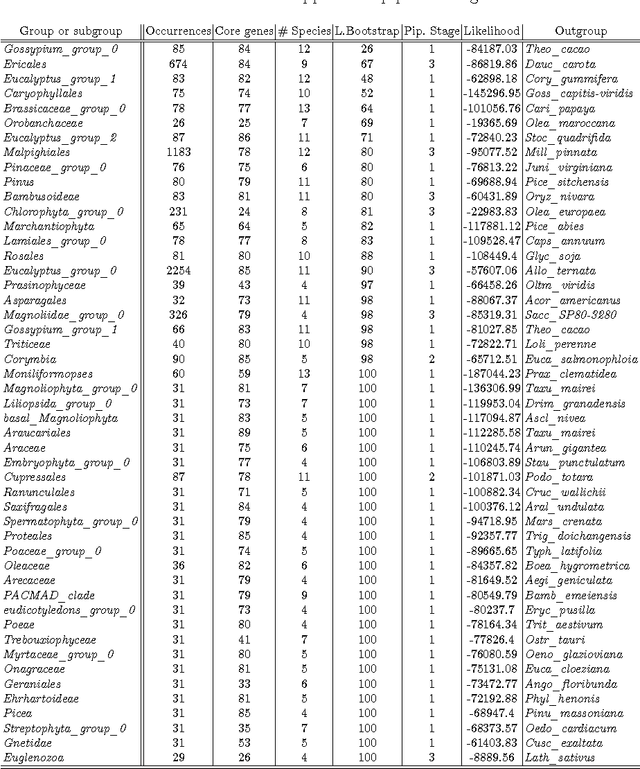
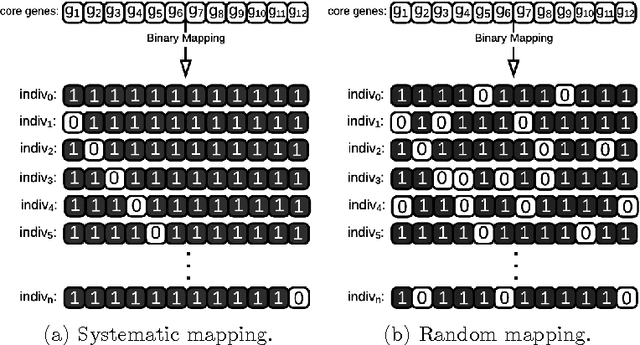
The amount of completely sequenced chloroplast genomes increases rapidly every day, leading to the possibility to build large scale phylogenetic trees of plant species. Considering a subset of close plant species defined according to their chloroplasts, the phylogenetic tree that can be inferred by their core genes is not necessarily well supported, due to the possible occurrence of "problematic" genes (i.e., homoplasy, incomplete lineage sorting, horizontal gene transfers, etc.) which may blur phylogenetic signal. However, a trustworthy phylogenetic tree can still be obtained if the number of problematic genes is low, the problem being to determine the largest subset of core genes that produces the best supported tree. To discard problematic genes and due to the overwhelming number of possible combinations, we propose an hybrid approach that embeds both genetic algorithms and statistical tests. Given a set of organisms, the result is a pipeline of many stages for the production of well supported phylogenetic trees. The proposal has been applied to different cases of plant families, leading to encouraging results for these families.
Gene Similarity-based Approaches for Determining Core-Genes of Chloroplasts
Dec 17, 2014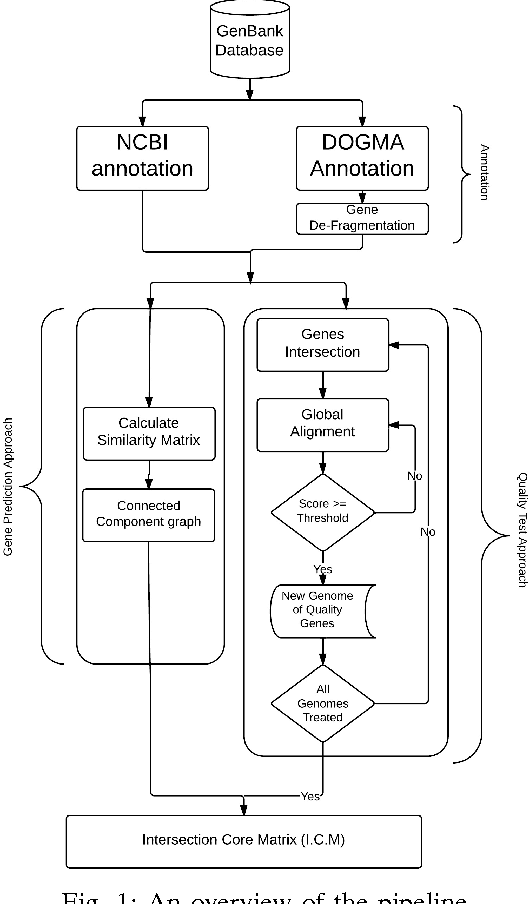
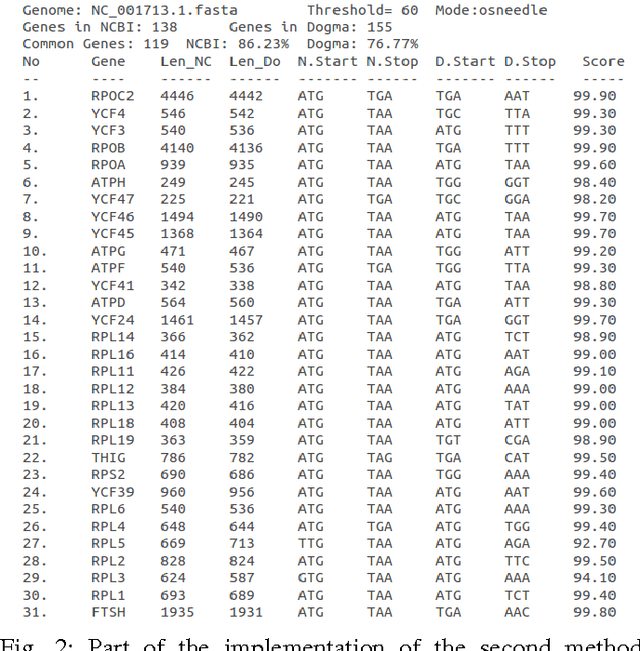
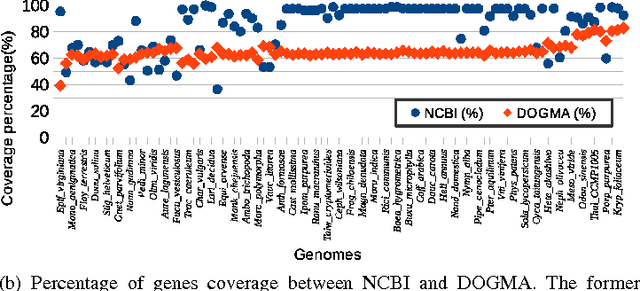
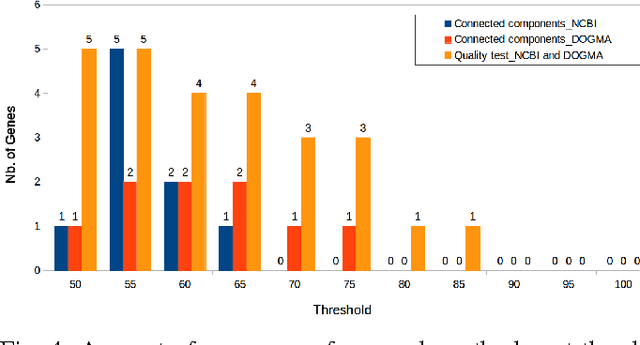
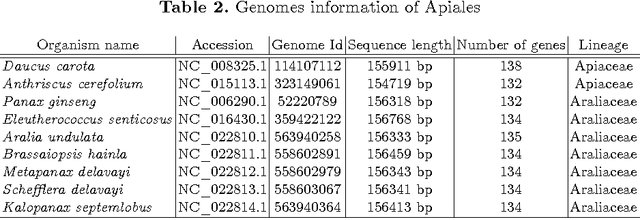
In computational biology and bioinformatics, the manner to understand evolution processes within various related organisms paid a lot of attention these last decades. However, accurate methodologies are still needed to discover genes content evolution. In a previous work, two novel approaches based on sequence similarities and genes features have been proposed. More precisely, we proposed to use genes names, sequence similarities, or both, insured either from NCBI or from DOGMA annotation tools. Dogma has the advantage to be an up-to-date accurate automatic tool specifically designed for chloroplasts, whereas NCBI possesses high quality human curated genes (together with wrongly annotated ones). The key idea of the former proposal was to take the best from these two tools. However, the first proposal was limited by name variations and spelling errors on the NCBI side, leading to core trees of low quality. In this paper, these flaws are fixed by improving the comparison of NCBI and DOGMA results, and by relaxing constraints on gene names while adding a stage of post-validation on gene sequences. The two stages of similarity measures, on names and sequences, are thus proposed for sequence clustering. This improves results that can be obtained using either NCBI or DOGMA alone. Results obtained with this quality control test are further investigated and compared with previously released ones, on both computational and biological aspects, considering a set of 99 chloroplastic genomes.