 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWell-supported phylogenies using largest subsets of core-genes by discrete particle swarm optimization
Jun 25, 2017

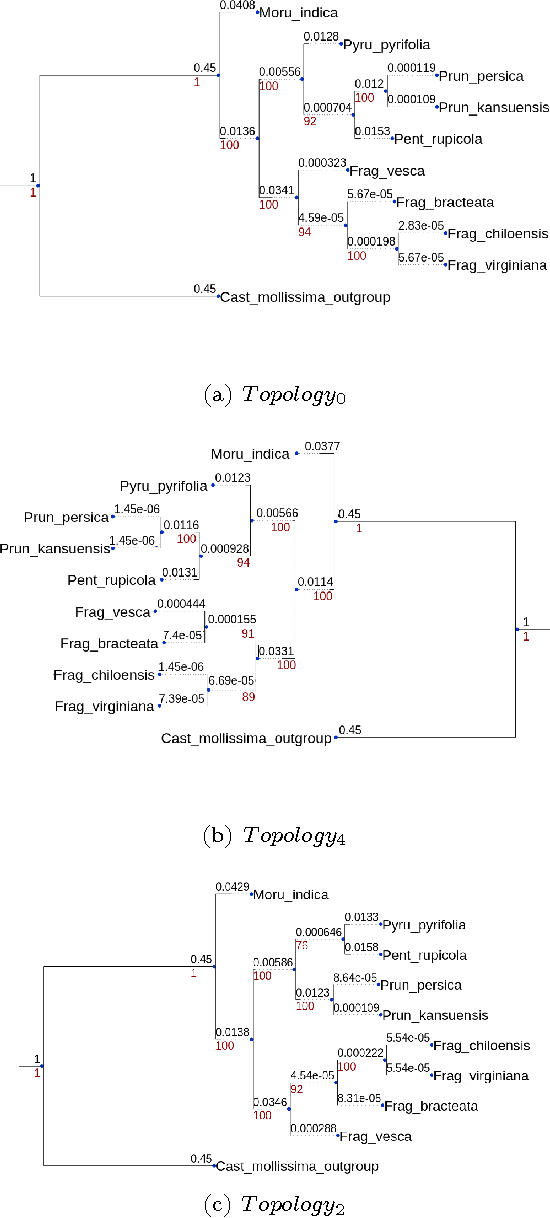
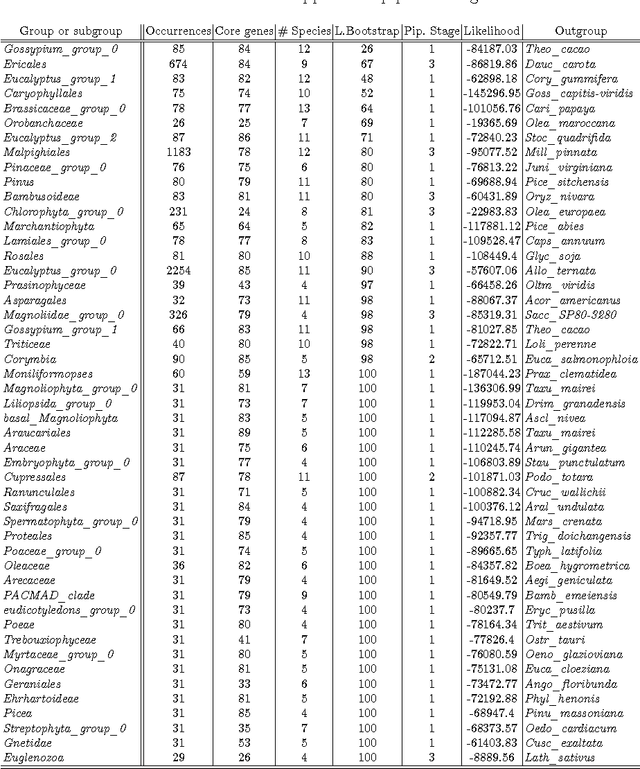
The number of complete chloroplastic genomes increases day after day, making it possible to rethink plants phylogeny at the biomolecular era. Given a set of close plants sharing in the order of one hundred of core chloroplastic genes, this article focuses on how to extract the largest subset of sequences in order to obtain the most supported species tree. Due to computational complexity, a discrete and distributed Particle Swarm Optimization (DPSO) is proposed. It is finally applied to the core genes of Rosales order.
Binary Particle Swarm Optimization versus Hybrid Genetic Algorithm for Inferring Well Supported Phylogenetic Trees
Aug 31, 2016
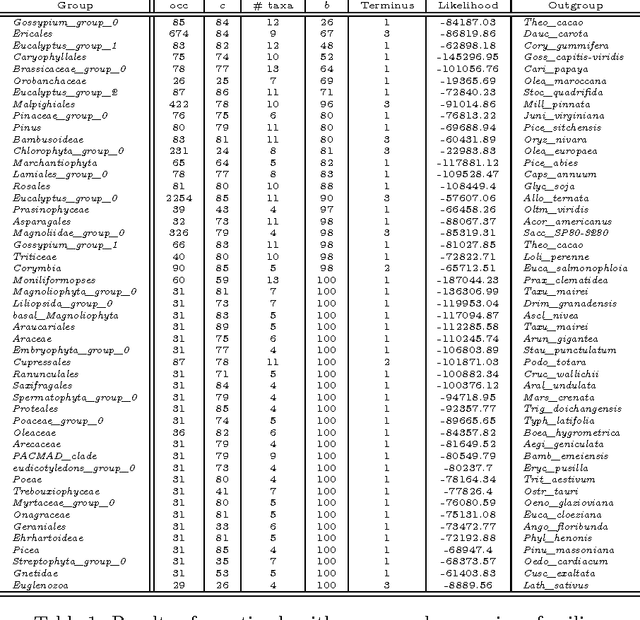

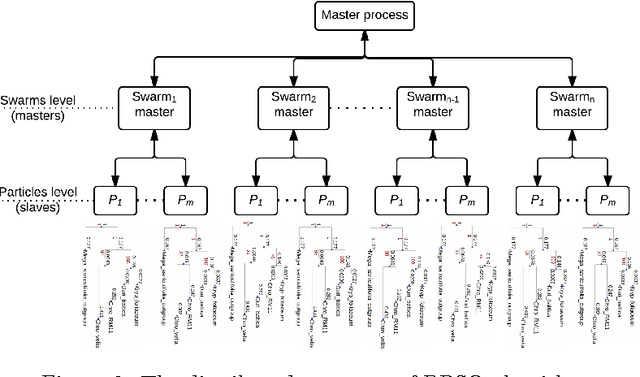
The amount of completely sequenced chloroplast genomes increases rapidly every day, leading to the possibility to build large-scale phylogenetic trees of plant species. Considering a subset of close plant species defined according to their chloroplasts, the phylogenetic tree that can be inferred by their core genes is not necessarily well supported, due to the possible occurrence of problematic genes (i.e., homoplasy, incomplete lineage sorting, horizontal gene transfers, etc.) which may blur the phylogenetic signal. However, a trustworthy phylogenetic tree can still be obtained provided such a number of blurring genes is reduced. The problem is thus to determine the largest subset of core genes that produces the best-supported tree. To discard problematic genes and due to the overwhelming number of possible combinations, this article focuses on how to extract the largest subset of sequences in order to obtain the most supported species tree. Due to computational complexity, a distributed Binary Particle Swarm Optimization (BPSO) is proposed in sequential and distributed fashions. Obtained results from both versions of the BPSO are compared with those computed using an hybrid approach embedding both genetic algorithms and statistical tests. The proposal has been applied to different cases of plant families, leading to encouraging results for these families.
Hybrid Genetic Algorithm and Lasso Test Approach for Inferring Well Supported Phylogenetic Trees based on Subsets of Chloroplastic Core Genes
Apr 20, 2015
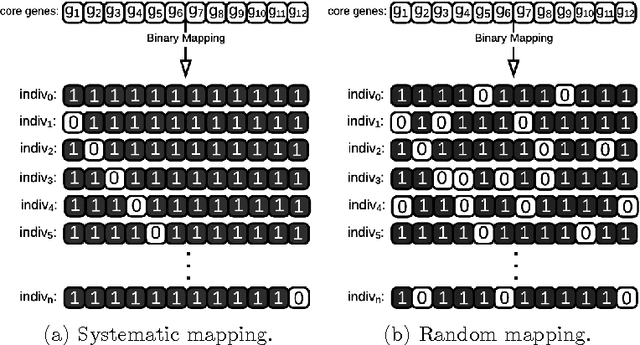
The amount of completely sequenced chloroplast genomes increases rapidly every day, leading to the possibility to build large scale phylogenetic trees of plant species. Considering a subset of close plant species defined according to their chloroplasts, the phylogenetic tree that can be inferred by their core genes is not necessarily well supported, due to the possible occurrence of "problematic" genes (i.e., homoplasy, incomplete lineage sorting, horizontal gene transfers, etc.) which may blur phylogenetic signal. However, a trustworthy phylogenetic tree can still be obtained if the number of problematic genes is low, the problem being to determine the largest subset of core genes that produces the best supported tree. To discard problematic genes and due to the overwhelming number of possible combinations, we propose an hybrid approach that embeds both genetic algorithms and statistical tests. Given a set of organisms, the result is a pipeline of many stages for the production of well supported phylogenetic trees. The proposal has been applied to different cases of plant families, leading to encouraging results for these families.
Gene Similarity-based Approaches for Determining Core-Genes of Chloroplasts
Dec 17, 2014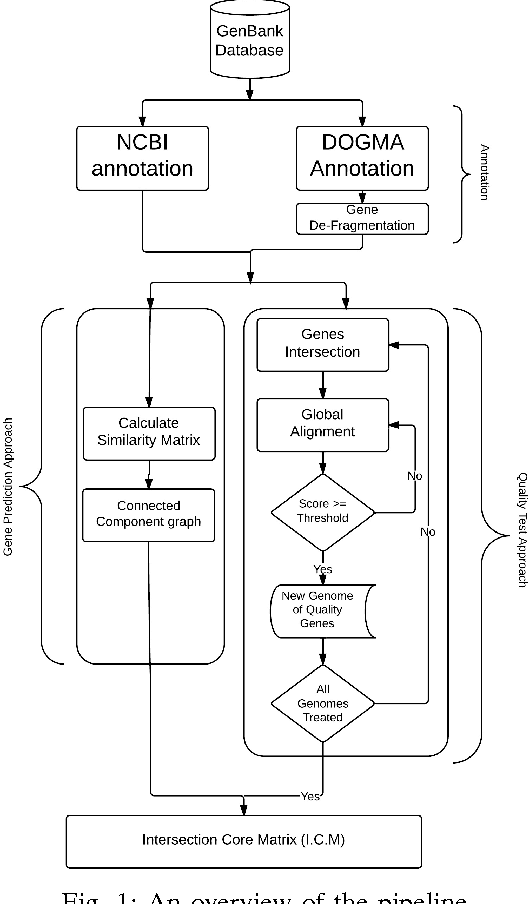
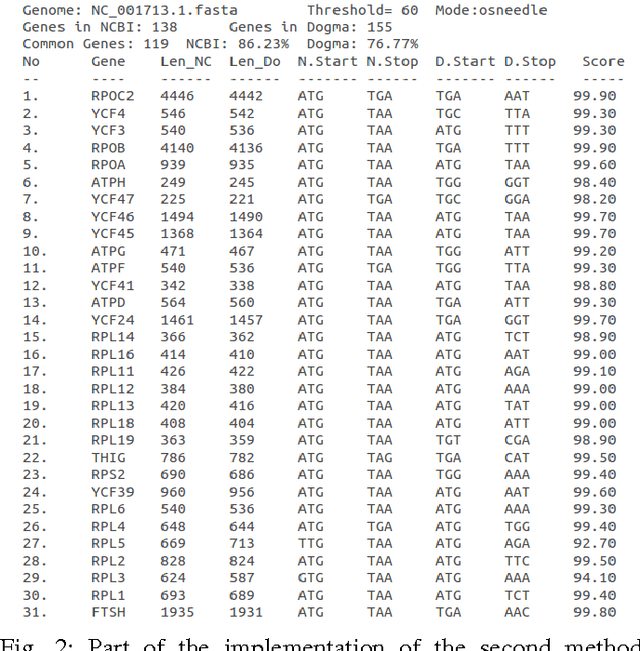
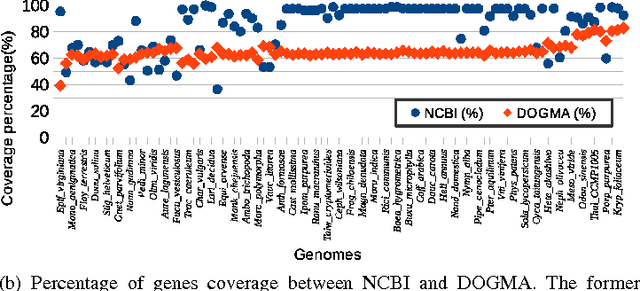
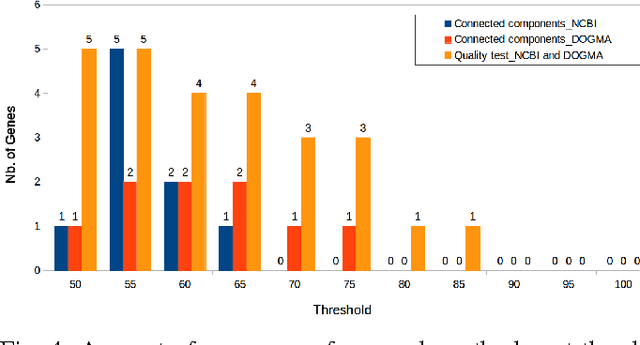
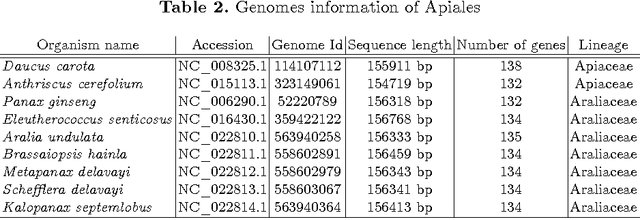
In computational biology and bioinformatics, the manner to understand evolution processes within various related organisms paid a lot of attention these last decades. However, accurate methodologies are still needed to discover genes content evolution. In a previous work, two novel approaches based on sequence similarities and genes features have been proposed. More precisely, we proposed to use genes names, sequence similarities, or both, insured either from NCBI or from DOGMA annotation tools. Dogma has the advantage to be an up-to-date accurate automatic tool specifically designed for chloroplasts, whereas NCBI possesses high quality human curated genes (together with wrongly annotated ones). The key idea of the former proposal was to take the best from these two tools. However, the first proposal was limited by name variations and spelling errors on the NCBI side, leading to core trees of low quality. In this paper, these flaws are fixed by improving the comparison of NCBI and DOGMA results, and by relaxing constraints on gene names while adding a stage of post-validation on gene sequences. The two stages of similarity measures, on names and sequences, are thus proposed for sequence clustering. This improves results that can be obtained using either NCBI or DOGMA alone. Results obtained with this quality control test are further investigated and compared with previously released ones, on both computational and biological aspects, considering a set of 99 chloroplastic genomes.