 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGene Similarity-based Approaches for Determining Core-Genes of Chloroplasts
Paper and Code
Dec 17, 2014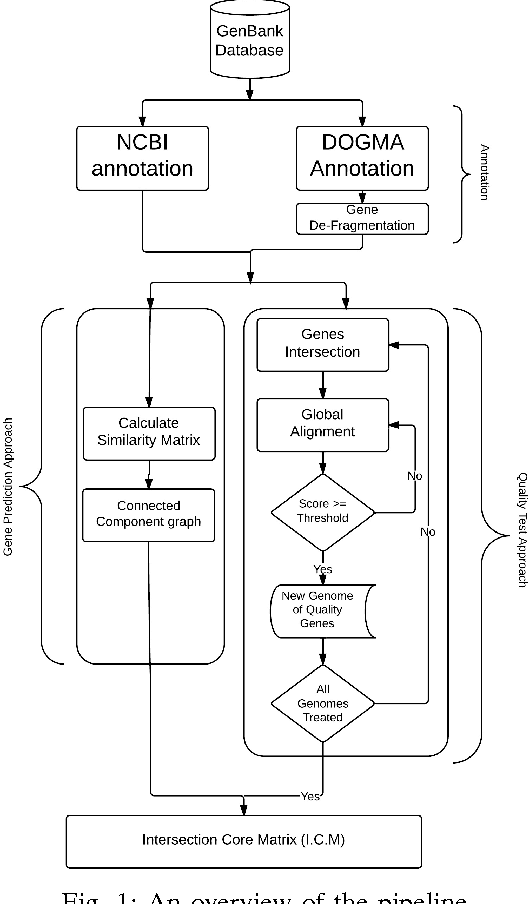
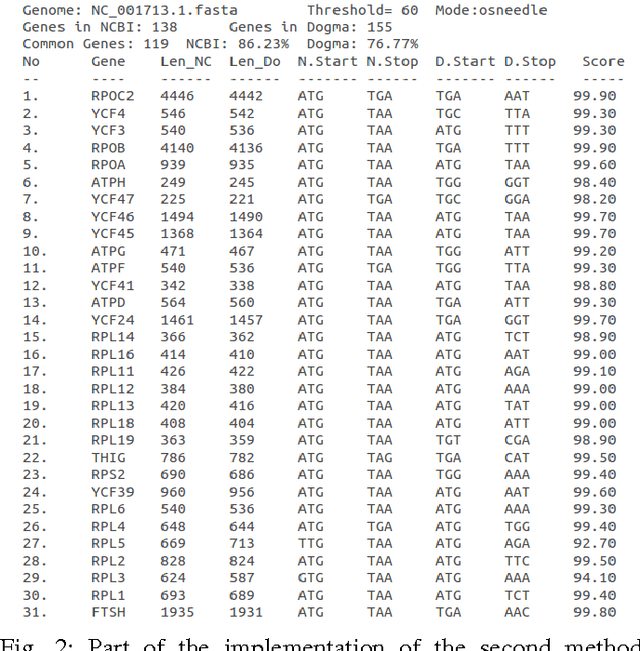
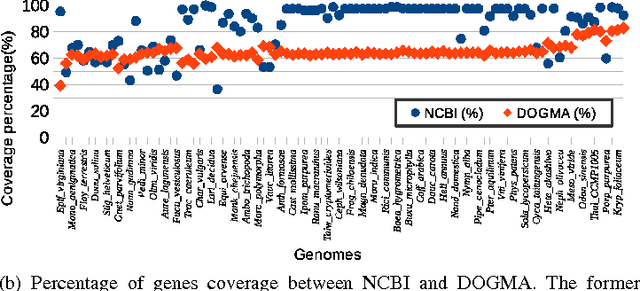
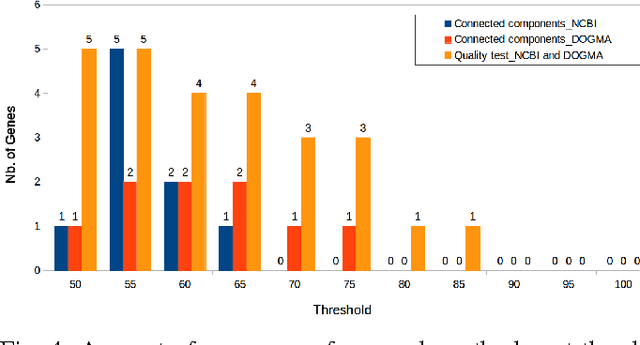
In computational biology and bioinformatics, the manner to understand evolution processes within various related organisms paid a lot of attention these last decades. However, accurate methodologies are still needed to discover genes content evolution. In a previous work, two novel approaches based on sequence similarities and genes features have been proposed. More precisely, we proposed to use genes names, sequence similarities, or both, insured either from NCBI or from DOGMA annotation tools. Dogma has the advantage to be an up-to-date accurate automatic tool specifically designed for chloroplasts, whereas NCBI possesses high quality human curated genes (together with wrongly annotated ones). The key idea of the former proposal was to take the best from these two tools. However, the first proposal was limited by name variations and spelling errors on the NCBI side, leading to core trees of low quality. In this paper, these flaws are fixed by improving the comparison of NCBI and DOGMA results, and by relaxing constraints on gene names while adding a stage of post-validation on gene sequences. The two stages of similarity measures, on names and sequences, are thus proposed for sequence clustering. This improves results that can be obtained using either NCBI or DOGMA alone. Results obtained with this quality control test are further investigated and compared with previously released ones, on both computational and biological aspects, considering a set of 99 chloroplastic genomes.