 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVerSe: Integrating Multiple Queries as Prompts for Versatile Cardiac MRI Segmentation
Dec 20, 2024Despite the advances in learning-based image segmentation approach, the accurate segmentation of cardiac structures from magnetic resonance imaging (MRI) remains a critical challenge. While existing automatic segmentation methods have shown promise, they still require extensive manual corrections of the segmentation results by human experts, particularly in complex regions such as the basal and apical parts of the heart. Recent efforts have been made on developing interactive image segmentation methods that enable human-in-the-loop learning. However, they are semi-automatic and inefficient, due to their reliance on click-based prompts, especially for 3D cardiac MRI volumes. To address these limitations, we propose VerSe, a Versatile Segmentation framework to unify automatic and interactive segmentation through mutiple queries. Our key innovation lies in the joint learning of object and click queries as prompts for a shared segmentation backbone. VerSe supports both fully automatic segmentation, through object queries, and interactive mask refinement, by providing click queries when needed. With the proposed integrated prompting scheme, VerSe demonstrates significant improvement in performance and efficiency over existing methods, on both cardiac MRI and out-of-distribution medical imaging datasets. The code is available at https://github.com/bangwayne/Verse.
Learning Volumetric Neural Deformable Models to Recover 3D Regional Heart Wall Motion from Multi-Planar Tagged MRI
Nov 21, 2024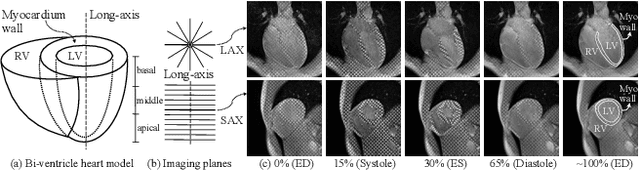
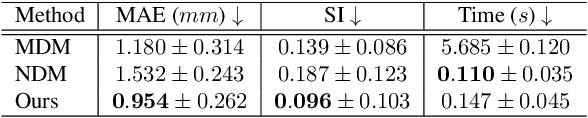
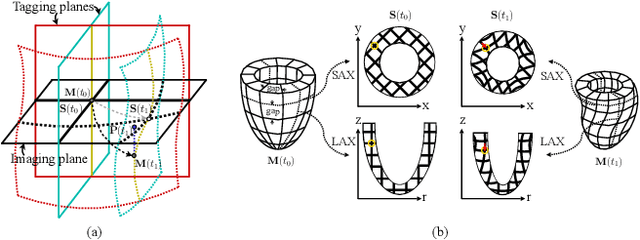
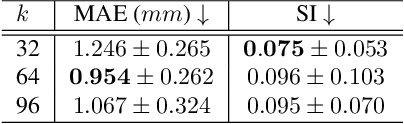
Multi-planar tagged MRI is the gold standard for regional heart wall motion evaluation. However, accurate recovery of the 3D true heart wall motion from a set of 2D apparent motion cues is challenging, due to incomplete sampling of the true motion and difficulty in information fusion from apparent motion cues observed on multiple imaging planes. To solve these challenges, we introduce a novel class of volumetric neural deformable models ($\upsilon$NDMs). Our $\upsilon$NDMs represent heart wall geometry and motion through a set of low-dimensional global deformation parameter functions and a diffeomorphic point flow regularized local deformation field. To learn such global and local deformation for 2D apparent motion mapping to 3D true motion, we design a hybrid point transformer, which incorporates both point cross-attention and self-attention mechanisms. While use of point cross-attention can learn to fuse 2D apparent motion cues into material point true motion hints, point self-attention hierarchically organised as an encoder-decoder structure can further learn to refine these hints and map them into 3D true motion. We have performed experiments on a large cohort of synthetic 3D regional heart wall motion dataset. The results demonstrated the high accuracy of our method for the recovery of dense 3D true motion from sparse 2D apparent motion cues. Project page is at https://github.com/DeepTag/VolumetricNeuralDeformableModels.
DPSeq: A Novel and Efficient Digital Pathology Classifier for Predicting Cancer Biomarkers using Sequencer Architecture
May 03, 2023In digital pathology tasks, transformers have achieved state-of-the-art results, surpassing convolutional neural networks (CNNs). However, transformers are usually complex and resource intensive. In this study, we developed a novel and efficient digital pathology classifier called DPSeq, to predict cancer biomarkers through fine-tuning a sequencer architecture integrating horizon and vertical bidirectional long short-term memory (BiLSTM) networks. Using hematoxylin and eosin (H&E)-stained histopathological images of colorectal cancer (CRC) from two international datasets: The Cancer Genome Atlas (TCGA) and Molecular and Cellular Oncology (MCO), the predictive performance of DPSeq was evaluated in series of experiments. DPSeq demonstrated exceptional performance for predicting key biomarkers in CRC (MSI status, Hypermutation, CIMP status, BRAF mutation, TP53 mutation and chromosomal instability [CING]), outperforming most published state-of-the-art classifiers in a within-cohort internal validation and a cross-cohort external validation. Additionally, under the same experimental conditions using the same set of training and testing datasets, DPSeq surpassed 4 CNN (ResNet18, ResNet50, MobileNetV2, and EfficientNet) and 2 transformer (ViT and Swin-T) models, achieving the highest AUROC and AUPRC values in predicting MSI status, BRAF mutation, and CIMP status. Furthermore, DPSeq required less time for both training and prediction due to its simple architecture. Therefore, DPSeq appears to be the preferred choice over transformer and CNN models for predicting cancer biomarkers.
Time to Embrace Natural Language Processing (NLP)-based Digital Pathology: Benchmarking NLP- and Convolutional Neural Network-based Deep Learning Pipelines
Feb 21, 2023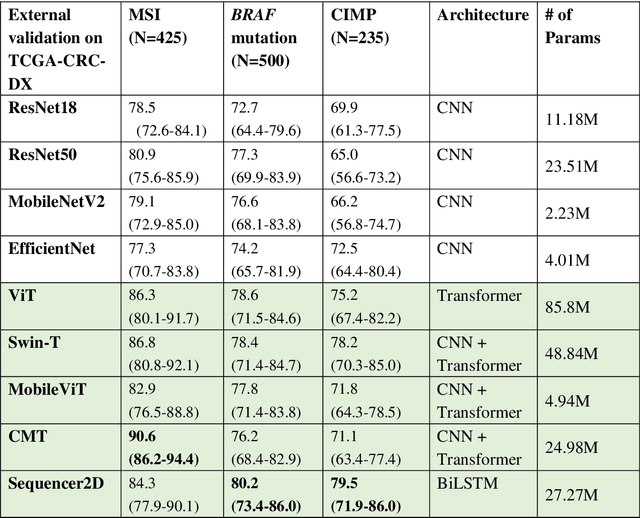
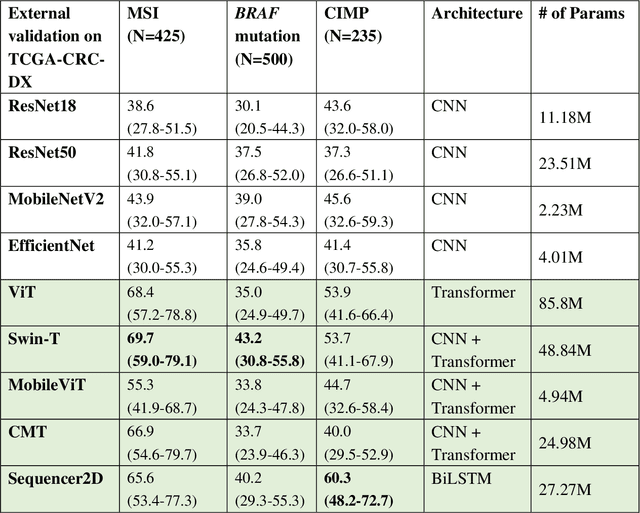
NLP-based computer vision models, particularly vision transformers, have been shown to outperform CNN models in many imaging tasks. However, most digital pathology artificial-intelligence models are based on CNN architectures, probably owing to a lack of data regarding NLP models for pathology images. In this study, we developed digital pathology pipelines to benchmark the five most recently proposed NLP models (vision transformer (ViT), Swin Transformer, MobileViT, CMT, and Sequencer2D) and four popular CNN models (ResNet18, ResNet50, MobileNetV2, and EfficientNet) to predict biomarkers in colorectal cancer (microsatellite instability, CpG island methylator phenotype, and BRAF mutation). Hematoxylin and eosin-stained whole-slide images from Molecular and Cellular Oncology and The Cancer Genome Atlas were used as training and external validation datasets, respectively. Cross-study external validations revealed that the NLP-based models significantly outperformed the CNN-based models in biomarker prediction tasks, improving the overall prediction and precision up to approximately 10% and 26%, respectively. Notably, compared with existing models in the current literature using large training datasets, our NLP models achieved state-of-the-art predictions for all three biomarkers using a relatively small training dataset, suggesting that large training datasets are not a prerequisite for NLP models or transformers, and NLP may be more suitable for clinical studies in which small training datasets are commonly collected. The superior performance of Sequencer2D suggests that further research and innovation on both transformer and bidirectional long short-term memory architectures are warranted in the field of digital pathology. NLP models can replace classic CNN architectures and become the new workhorse backbone in the field of digital pathology.
Predicting microsatellite instability and key biomarkers in colorectal cancer from H&E-stained images: Achieving SOTA with Less Data using Swin Transformer
Aug 22, 2022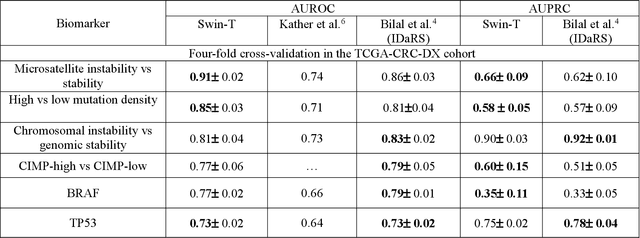
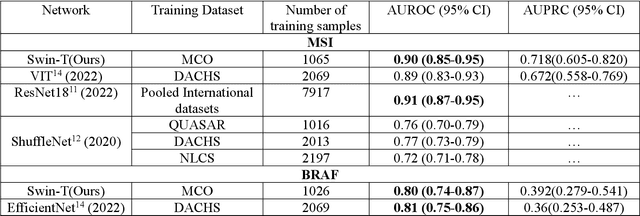
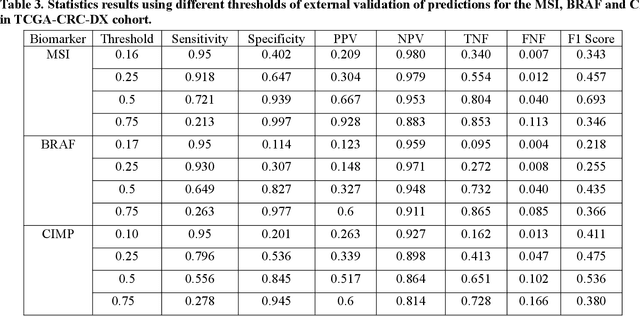
Artificial intelligence (AI) models have been developed for predicting clinically relevant biomarkers, including microsatellite instability (MSI), for colorectal cancers (CRC). However, the current deep-learning networks are data-hungry and require large training datasets, which are often lacking in the medical domain. In this study, based on the latest Hierarchical Vision Transformer using Shifted Windows (Swin-T), we developed an efficient workflow for biomarkers in CRC (MSI, hypermutation, chromosomal instability, CpG island methylator phenotype, BRAF, and TP53 mutation) that only required relatively small datasets, but achieved the state-of-the-art (SOTA) predictive performance. Our Swin-T workflow not only substantially outperformed published models in an intra-study cross-validation experiment using TCGA-CRC-DX dataset (N = 462), but also showed excellent generalizability in cross-study external validation and delivered a SOTA AUROC of 0.90 for MSI using the MCO dataset for training (N = 1065) and the same TCGA-CRC-DX for testing. Similar performance (AUROC=0.91) was achieved by Echle and colleagues using 8000 training samples (ResNet18) on the same testing dataset. Swin-T was extremely efficient using small training datasets and exhibits robust predictive performance with only 200-500 training samples. These data indicate that Swin-T may be 5-10 times more efficient than the current state-of-the-art algorithms for MSI based on ResNet18 and ShuffleNet. Furthermore, the Swin-T models showed promise as pre-screening tests for MSI status and BRAF mutation status, which could exclude and reduce the samples before the subsequent standard testing in a cascading diagnostic workflow to allow turnaround time reduction and cost saving.
A robust and lightweight deep attention multiple instance learning algorithm for predicting genetic alterations
May 31, 2022Deep-learning models based on whole-slide digital pathology images (WSIs) become increasingly popular for predicting molecular biomarkers. Instance-based models has been the mainstream strategy for predicting genetic alterations using WSIs although bag-based models along with self-attention mechanism-based algorithms have been proposed for other digital pathology applications. In this paper, we proposed a novel Attention-based Multiple Instance Mutation Learning (AMIML) model for predicting gene mutations. AMIML was comprised of successive 1-D convolutional layers, a decoder, and a residual weight connection to facilitate further integration of a lightweight attention mechanism to detect the most predictive image patches. Using data for 24 clinically relevant genes from four cancer cohorts in The Cancer Genome Atlas (TCGA) studies (UCEC, BRCA, GBM and KIRC), we compared AMIML with one popular instance-based model and four recently published bag-based models (e.g., CHOWDER, HE2RNA, etc.). AMIML demonstrated excellent robustness, not only outperforming all the five baseline algorithms in the vast majority of the tested genes (17 out of 24), but also providing near-best-performance for the other seven genes. Conversely, the performance of the baseline published algorithms varied across different cancers/genes. In addition, compared to the published models for genetic alterations, AMIML provided a significant improvement for predicting a wide range of genes (e.g., KMT2C, TP53, and SETD2 for KIRC; ERBB2, BRCA1, and BRCA2 for BRCA; JAK1, POLE, and MTOR for UCEC) as well as produced outstanding predictive models for other clinically relevant gene mutations, which have not been reported in the current literature. Furthermore, with the flexible and interpretable attention-based MIL pooling mechanism, AMIML could further zero-in and detect predictive image patches.