 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDPSeq: A Novel and Efficient Digital Pathology Classifier for Predicting Cancer Biomarkers using Sequencer Architecture
May 03, 2023In digital pathology tasks, transformers have achieved state-of-the-art results, surpassing convolutional neural networks (CNNs). However, transformers are usually complex and resource intensive. In this study, we developed a novel and efficient digital pathology classifier called DPSeq, to predict cancer biomarkers through fine-tuning a sequencer architecture integrating horizon and vertical bidirectional long short-term memory (BiLSTM) networks. Using hematoxylin and eosin (H&E)-stained histopathological images of colorectal cancer (CRC) from two international datasets: The Cancer Genome Atlas (TCGA) and Molecular and Cellular Oncology (MCO), the predictive performance of DPSeq was evaluated in series of experiments. DPSeq demonstrated exceptional performance for predicting key biomarkers in CRC (MSI status, Hypermutation, CIMP status, BRAF mutation, TP53 mutation and chromosomal instability [CING]), outperforming most published state-of-the-art classifiers in a within-cohort internal validation and a cross-cohort external validation. Additionally, under the same experimental conditions using the same set of training and testing datasets, DPSeq surpassed 4 CNN (ResNet18, ResNet50, MobileNetV2, and EfficientNet) and 2 transformer (ViT and Swin-T) models, achieving the highest AUROC and AUPRC values in predicting MSI status, BRAF mutation, and CIMP status. Furthermore, DPSeq required less time for both training and prediction due to its simple architecture. Therefore, DPSeq appears to be the preferred choice over transformer and CNN models for predicting cancer biomarkers.
Time to Embrace Natural Language Processing (NLP)-based Digital Pathology: Benchmarking NLP- and Convolutional Neural Network-based Deep Learning Pipelines
Feb 21, 2023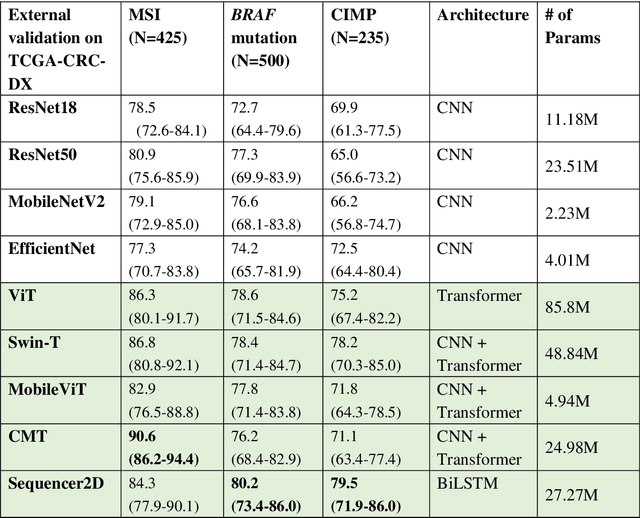
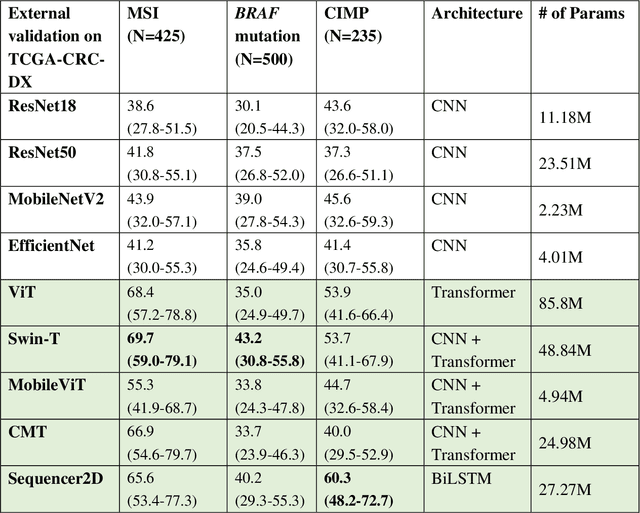
NLP-based computer vision models, particularly vision transformers, have been shown to outperform CNN models in many imaging tasks. However, most digital pathology artificial-intelligence models are based on CNN architectures, probably owing to a lack of data regarding NLP models for pathology images. In this study, we developed digital pathology pipelines to benchmark the five most recently proposed NLP models (vision transformer (ViT), Swin Transformer, MobileViT, CMT, and Sequencer2D) and four popular CNN models (ResNet18, ResNet50, MobileNetV2, and EfficientNet) to predict biomarkers in colorectal cancer (microsatellite instability, CpG island methylator phenotype, and BRAF mutation). Hematoxylin and eosin-stained whole-slide images from Molecular and Cellular Oncology and The Cancer Genome Atlas were used as training and external validation datasets, respectively. Cross-study external validations revealed that the NLP-based models significantly outperformed the CNN-based models in biomarker prediction tasks, improving the overall prediction and precision up to approximately 10% and 26%, respectively. Notably, compared with existing models in the current literature using large training datasets, our NLP models achieved state-of-the-art predictions for all three biomarkers using a relatively small training dataset, suggesting that large training datasets are not a prerequisite for NLP models or transformers, and NLP may be more suitable for clinical studies in which small training datasets are commonly collected. The superior performance of Sequencer2D suggests that further research and innovation on both transformer and bidirectional long short-term memory architectures are warranted in the field of digital pathology. NLP models can replace classic CNN architectures and become the new workhorse backbone in the field of digital pathology.
Predicting microsatellite instability and key biomarkers in colorectal cancer from H&E-stained images: Achieving SOTA with Less Data using Swin Transformer
Aug 22, 2022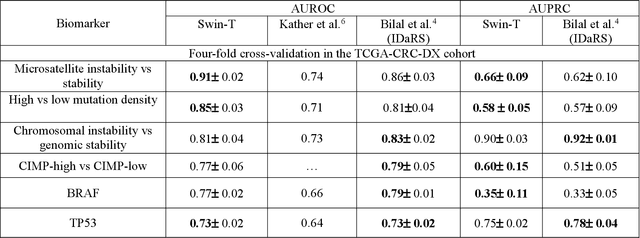
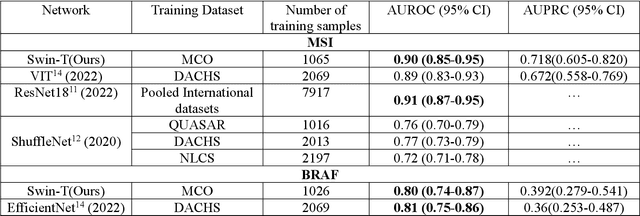
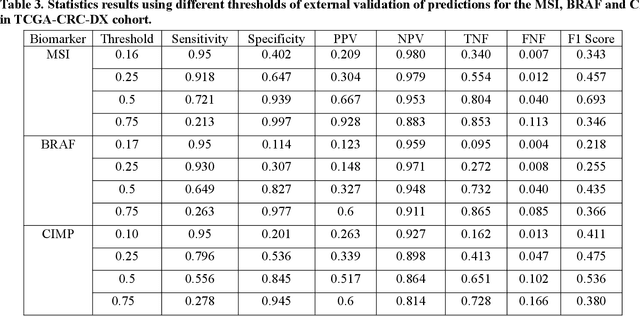
Artificial intelligence (AI) models have been developed for predicting clinically relevant biomarkers, including microsatellite instability (MSI), for colorectal cancers (CRC). However, the current deep-learning networks are data-hungry and require large training datasets, which are often lacking in the medical domain. In this study, based on the latest Hierarchical Vision Transformer using Shifted Windows (Swin-T), we developed an efficient workflow for biomarkers in CRC (MSI, hypermutation, chromosomal instability, CpG island methylator phenotype, BRAF, and TP53 mutation) that only required relatively small datasets, but achieved the state-of-the-art (SOTA) predictive performance. Our Swin-T workflow not only substantially outperformed published models in an intra-study cross-validation experiment using TCGA-CRC-DX dataset (N = 462), but also showed excellent generalizability in cross-study external validation and delivered a SOTA AUROC of 0.90 for MSI using the MCO dataset for training (N = 1065) and the same TCGA-CRC-DX for testing. Similar performance (AUROC=0.91) was achieved by Echle and colleagues using 8000 training samples (ResNet18) on the same testing dataset. Swin-T was extremely efficient using small training datasets and exhibits robust predictive performance with only 200-500 training samples. These data indicate that Swin-T may be 5-10 times more efficient than the current state-of-the-art algorithms for MSI based on ResNet18 and ShuffleNet. Furthermore, the Swin-T models showed promise as pre-screening tests for MSI status and BRAF mutation status, which could exclude and reduce the samples before the subsequent standard testing in a cascading diagnostic workflow to allow turnaround time reduction and cost saving.
A robust and lightweight deep attention multiple instance learning algorithm for predicting genetic alterations
May 31, 2022Deep-learning models based on whole-slide digital pathology images (WSIs) become increasingly popular for predicting molecular biomarkers. Instance-based models has been the mainstream strategy for predicting genetic alterations using WSIs although bag-based models along with self-attention mechanism-based algorithms have been proposed for other digital pathology applications. In this paper, we proposed a novel Attention-based Multiple Instance Mutation Learning (AMIML) model for predicting gene mutations. AMIML was comprised of successive 1-D convolutional layers, a decoder, and a residual weight connection to facilitate further integration of a lightweight attention mechanism to detect the most predictive image patches. Using data for 24 clinically relevant genes from four cancer cohorts in The Cancer Genome Atlas (TCGA) studies (UCEC, BRCA, GBM and KIRC), we compared AMIML with one popular instance-based model and four recently published bag-based models (e.g., CHOWDER, HE2RNA, etc.). AMIML demonstrated excellent robustness, not only outperforming all the five baseline algorithms in the vast majority of the tested genes (17 out of 24), but also providing near-best-performance for the other seven genes. Conversely, the performance of the baseline published algorithms varied across different cancers/genes. In addition, compared to the published models for genetic alterations, AMIML provided a significant improvement for predicting a wide range of genes (e.g., KMT2C, TP53, and SETD2 for KIRC; ERBB2, BRCA1, and BRCA2 for BRCA; JAK1, POLE, and MTOR for UCEC) as well as produced outstanding predictive models for other clinically relevant gene mutations, which have not been reported in the current literature. Furthermore, with the flexible and interpretable attention-based MIL pooling mechanism, AMIML could further zero-in and detect predictive image patches.
Colorectal cancer survival prediction using deep distribution based multiple-instance learning
Apr 24, 2022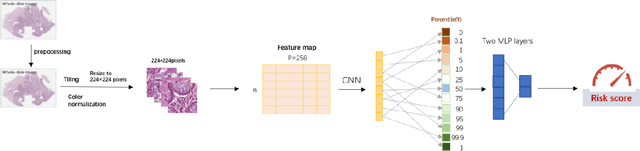
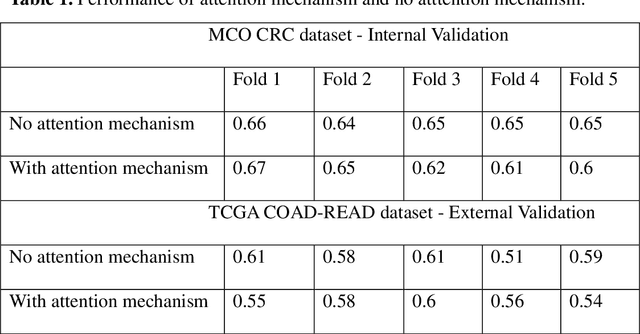
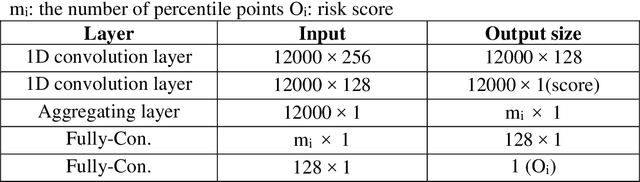
Several deep learning algorithms have been developed to predict survival of cancer patients using whole slide images (WSIs).However, identification of image phenotypes within the WSIs that are relevant to patient survival and disease progression is difficult for both clinicians, and deep learning algorithms. Most deep learning based Multiple Instance Learning (MIL) algorithms for survival prediction use either top instances (e.g., maxpooling) or top/bottom instances (e.g., MesoNet) to identify image phenotypes. In this study, we hypothesize that wholistic information of the distribution of the patch scores within a WSI can predict the cancer survival better. We developed a distribution based multiple-instance survival learning algorithm (DeepDisMISL) to validate this hypothesis. We designed and executed experiments using two large international colorectal cancer WSIs datasets - MCO CRC and TCGA COAD-READ. Our results suggest that the more information about the distribution of the patch scores for a WSI, the better is the prediction performance. Including multiple neighborhood instances around each selected distribution location (e.g., percentiles) could further improve the prediction. DeepDisMISL demonstrated superior predictive ability compared to other recently published, state-of-the-art algorithms. Furthermore, our algorithm is interpretable and could assist in understanding the relationship between cancer morphological phenotypes and patients cancer survival risk.
Optimize Deep Learning Models for Prediction of Gene Mutations Using Unsupervised Clustering
Mar 31, 2022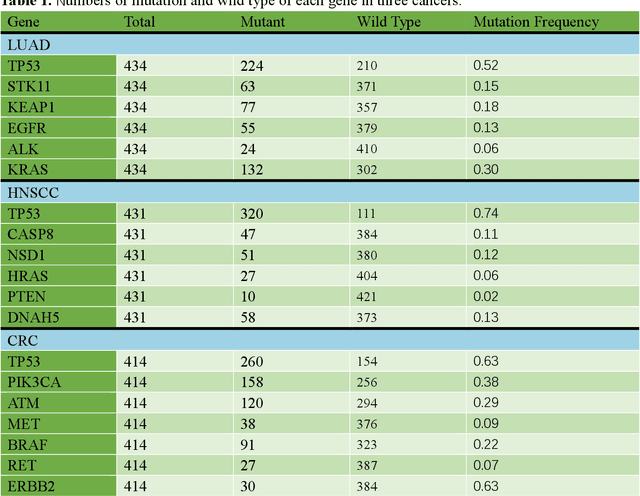
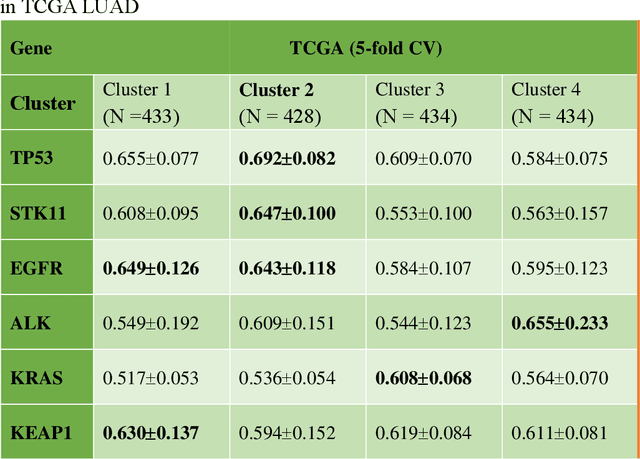
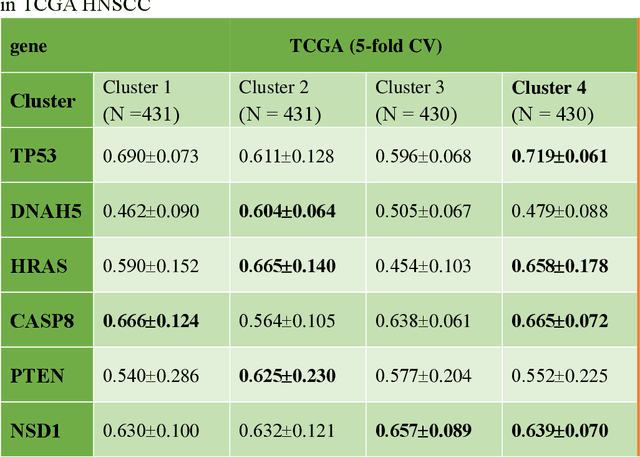
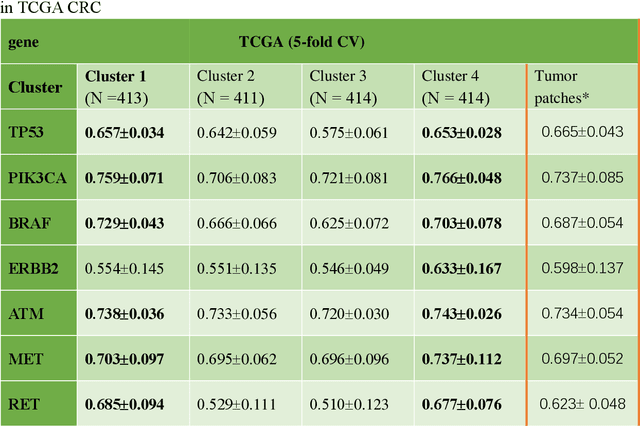
Deep learning has become the mainstream methodological choice for analyzing and interpreting whole-slide digital pathology images (WSIs). It is commonly assumed that tumor regions carry most predictive information. In this paper, we proposed an unsupervised clustering-based multiple-instance learning, and apply our method to develop deep-learning models for prediction of gene mutations using WSIs from three cancer types in The Cancer Genome Atlas (TCGA) studies (CRC, LUAD, and HNSCC). We showed that unsupervised clustering of image patches could help identify predictive patches, exclude patches lack of predictive information, and therefore improve prediction on gene mutations in all three different cancer types, compared with the WSI based method without selection of image patches and models based on only tumor regions. Additionally, our proposed algorithm outperformed two recently published baseline algorithms leveraging unsupervised clustering to assist model prediction. The unsupervised-clustering-based approach for mutation prediction allows identification of the spatial regions related to mutation of a specific gene via the resolved probability scores, highlighting the heterogeneity of a predicted genotype in the tumor microenvironment. Finally, our study also demonstrated that selection of tumor regions of WSIs is not always the best way to identify patches for prediction of gene mutations, and other tissue types in the tumor micro-environment may provide better prediction ability for gene mutations than tumor tissues.
Improving Feature Extraction from Histopathological Images Through A Fine-tuning ImageNet Model
Jan 03, 2022Due to lack of annotated pathological images, transfer learning has been the predominant approach in the field of digital pathology.Pre-trained neural networks based on ImageNet database are often used to extract "off the shelf" features, achieving great success in predicting tissue types, molecular features, and clinical outcomes, etc. We hypothesize that fine-tuning the pre-trained models using histopathological images could further improve feature extraction, and downstream prediction performance.We used 100,000 annotated HE image patches for colorectal cancer (CRC) to finetune a pretrained Xception model via a twostep approach.The features extracted from finetuned Xception (FTX2048) model and Imagepretrained (IMGNET2048) model were compared through: (1) tissue classification for HE images from CRC, same image type that was used for finetuning; (2) prediction of immunerelated gene expression and (3) gene mutations for lung adenocarcinoma (LUAD).Fivefold cross validation was used for model performance evaluation. The extracted features from the finetuned FTX2048 exhibited significantly higher accuracy for predicting tisue types of CRC compared to the off the shelf feature directly from Xception based on ImageNet database. Particularly, FTX2048 markedly improved the accuracy for stroma from 87% to 94%. Similarly, features from FTX2048 boosted the prediction of transcriptomic expression of immunerelated genesin LUAD. For the genes that had signigicant relationships with image fetures, the features fgrom the finetuned model imprroved the prediction for the majority of the genes. Inaddition, fetures from FTX2048 improved prediction of mutation for 5 out of 9 most frequently mutated genes in LUAD.
A Retrospective Analysis using Deep-Learning Models for Prediction of Survival Outcome and Benefit of Adjuvant Chemotherapy in Stage II/III Colorectal Cancer
Nov 05, 2021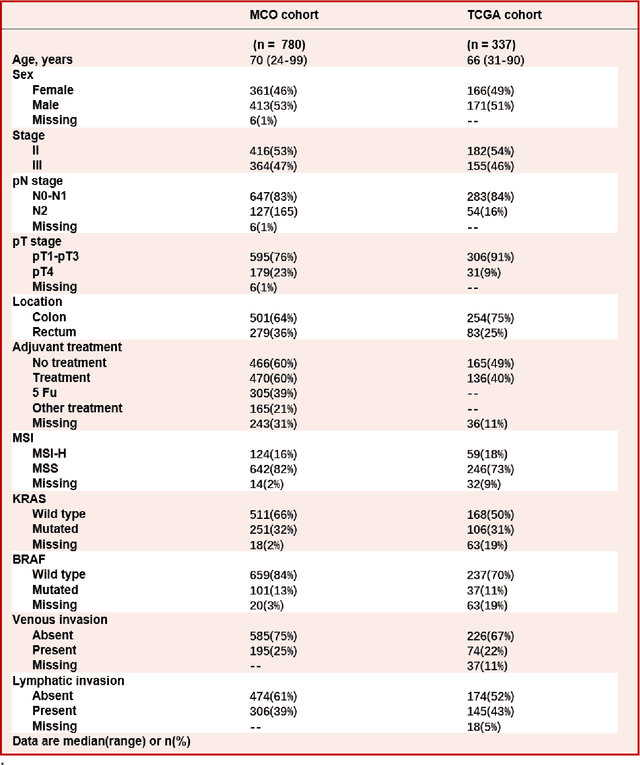
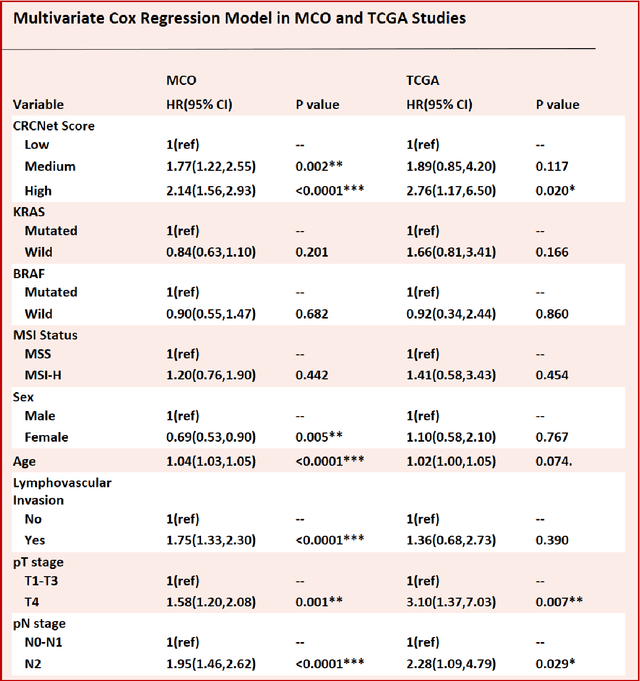
Most early-stage colorectal cancer (CRC) patients can be cured by surgery alone, and only certain high-risk early-stage CRC patients benefit from adjuvant chemotherapies. However, very few validated biomarkers are available to accurately predict survival benefit from postoperative chemotherapy. We developed a novel deep-learning algorithm (CRCNet) using whole-slide images from Molecular and Cellular Oncology (MCO) to predict survival benefit of adjuvant chemotherapy in stage II/III CRC. We validated CRCNet both internally through cross-validation and externally using an independent cohort from The Cancer Genome Atlas (TCGA). We showed that CRCNet can accurately predict not only survival prognosis but also the treatment effect of adjuvant chemotherapy. The CRCNet identified high-risk subgroup benefits from adjuvant chemotherapy most and significant longer survival is observed among chemo-treated patients. Conversely, minimal chemotherapy benefit is observed in the CRCNet low- and medium-risk subgroups. Therefore, CRCNet can potentially be of great use in guiding treatments for Stage II/III CRC.