 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAge Estimation Based on Graph Convolutional Networks and Multi-head Attention Mechanisms
Oct 12, 2023



Age estimation technology is a part of facial recognition and has been applied to identity authentication. This technology achieves the development and application of a juvenile anti-addiction system by authenticating users in the game. Convolutional Neural Network (CNN) and Transformer algorithms are widely used in this application scenario. However, these two models cannot flexibly extract and model features of faces with irregular shapes, and they are ineffective in capturing key information. Furthermore, the above methods will contain a lot of background information while extracting features, which will interfere with the model. In consequence, it is easy to extract redundant information from images. In this paper, a new modeling idea is proposed to solve this problem, which can flexibly model irregular objects. The Graph Convolutional Network (GCN) is used to extract features from irregular face images effectively, and multi-head attention mechanisms are added to avoid redundant features and capture key region information in the image. This model can effectively improve the accuracy of age estimation and reduce the MAE error value to about 3.64, which is better than the effect of today's age estimation model, to improve the accuracy of face recognition and identity authentication.
A robust and lightweight deep attention multiple instance learning algorithm for predicting genetic alterations
May 31, 2022Deep-learning models based on whole-slide digital pathology images (WSIs) become increasingly popular for predicting molecular biomarkers. Instance-based models has been the mainstream strategy for predicting genetic alterations using WSIs although bag-based models along with self-attention mechanism-based algorithms have been proposed for other digital pathology applications. In this paper, we proposed a novel Attention-based Multiple Instance Mutation Learning (AMIML) model for predicting gene mutations. AMIML was comprised of successive 1-D convolutional layers, a decoder, and a residual weight connection to facilitate further integration of a lightweight attention mechanism to detect the most predictive image patches. Using data for 24 clinically relevant genes from four cancer cohorts in The Cancer Genome Atlas (TCGA) studies (UCEC, BRCA, GBM and KIRC), we compared AMIML with one popular instance-based model and four recently published bag-based models (e.g., CHOWDER, HE2RNA, etc.). AMIML demonstrated excellent robustness, not only outperforming all the five baseline algorithms in the vast majority of the tested genes (17 out of 24), but also providing near-best-performance for the other seven genes. Conversely, the performance of the baseline published algorithms varied across different cancers/genes. In addition, compared to the published models for genetic alterations, AMIML provided a significant improvement for predicting a wide range of genes (e.g., KMT2C, TP53, and SETD2 for KIRC; ERBB2, BRCA1, and BRCA2 for BRCA; JAK1, POLE, and MTOR for UCEC) as well as produced outstanding predictive models for other clinically relevant gene mutations, which have not been reported in the current literature. Furthermore, with the flexible and interpretable attention-based MIL pooling mechanism, AMIML could further zero-in and detect predictive image patches.
Optimize Deep Learning Models for Prediction of Gene Mutations Using Unsupervised Clustering
Mar 31, 2022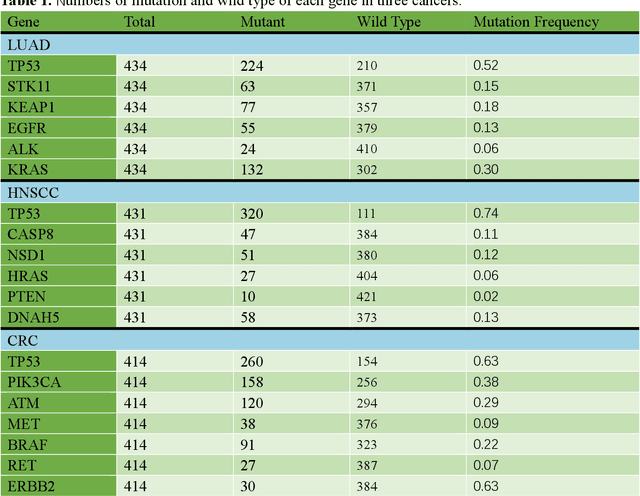
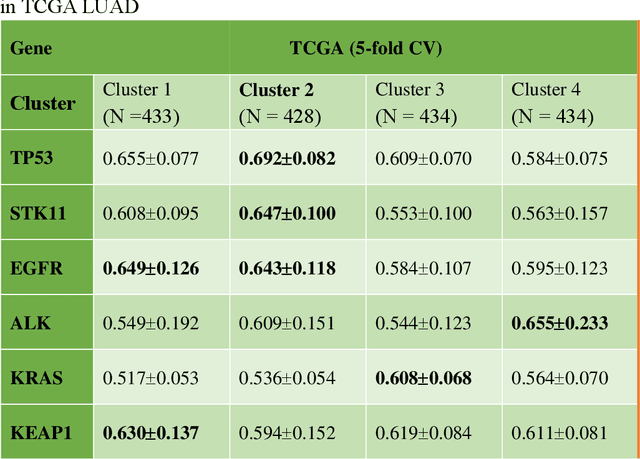
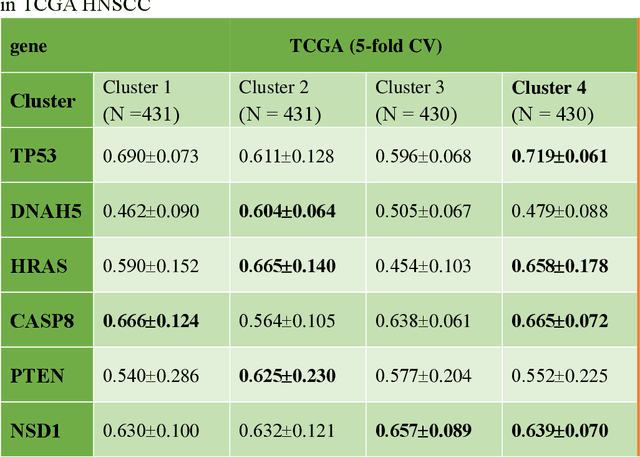
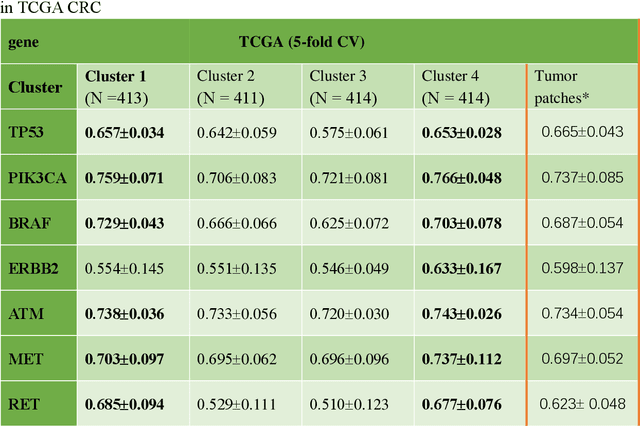
Deep learning has become the mainstream methodological choice for analyzing and interpreting whole-slide digital pathology images (WSIs). It is commonly assumed that tumor regions carry most predictive information. In this paper, we proposed an unsupervised clustering-based multiple-instance learning, and apply our method to develop deep-learning models for prediction of gene mutations using WSIs from three cancer types in The Cancer Genome Atlas (TCGA) studies (CRC, LUAD, and HNSCC). We showed that unsupervised clustering of image patches could help identify predictive patches, exclude patches lack of predictive information, and therefore improve prediction on gene mutations in all three different cancer types, compared with the WSI based method without selection of image patches and models based on only tumor regions. Additionally, our proposed algorithm outperformed two recently published baseline algorithms leveraging unsupervised clustering to assist model prediction. The unsupervised-clustering-based approach for mutation prediction allows identification of the spatial regions related to mutation of a specific gene via the resolved probability scores, highlighting the heterogeneity of a predicted genotype in the tumor microenvironment. Finally, our study also demonstrated that selection of tumor regions of WSIs is not always the best way to identify patches for prediction of gene mutations, and other tissue types in the tumor micro-environment may provide better prediction ability for gene mutations than tumor tissues.