 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeadabmDCA 2.0 -- a flexible but easy-to-use package for Direct Coupling Analysis
Jan 30, 2025In this methods article, we provide a flexible but easy-to-use implementation of Direct Coupling Analysis (DCA) based on Boltzmann machine learning, together with a tutorial on how to use it. The package \texttt{adabmDCA 2.0} is available in different programming languages (C++, Julia, Python) usable on different architectures (single-core and multi-core CPU, GPU) using a common front-end interface. In addition to several learning protocols for dense and sparse generative DCA models, it allows to directly address common downstream tasks like residue-residue contact prediction, mutational-effect prediction, scoring of sequence libraries and generation of artificial sequences for sequence design. It is readily applicable to protein and RNA sequence data.
Inference in conditioned dynamics through causality restoration
Oct 18, 2022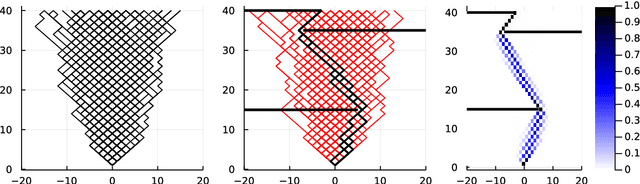
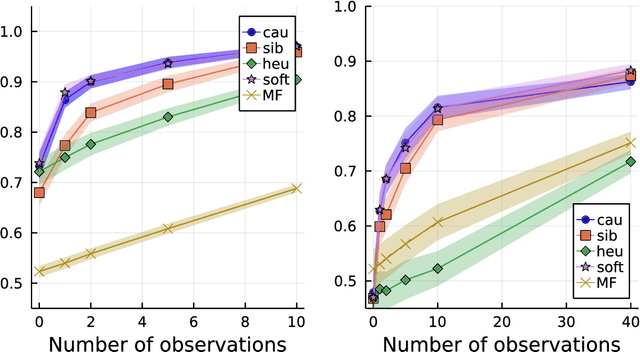
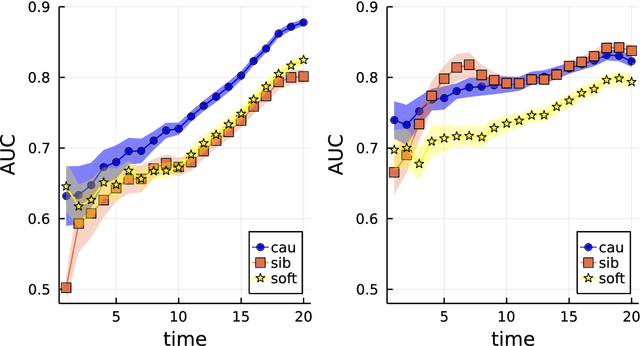
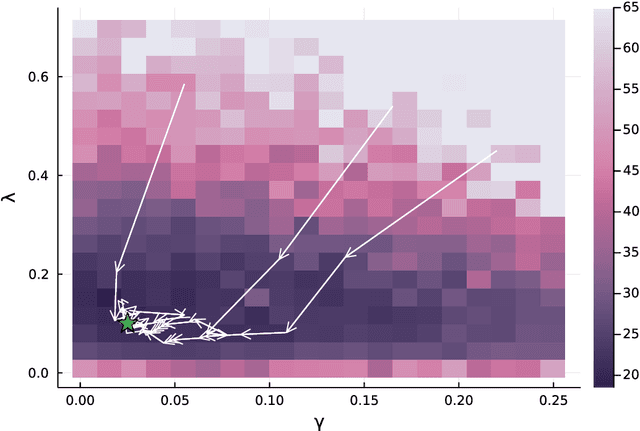
Computing observables from conditioned dynamics is typically computationally hard, because, although obtaining independent samples efficiently from the unconditioned dynamics is usually feasible, generally most of the samples must be discarded (in a form of importance sampling) because they do not satisfy the imposed conditions. Sampling directly from the conditioned distribution is non-trivial, as conditioning breaks the causal properties of the dynamics which ultimately renders the sampling procedure efficient. One standard way of achieving it is through a Metropolis Monte-Carlo procedure, but this procedure is normally slow and a very large number of Monte-Carlo steps is needed to obtain a small number of statistically independent samples. In this work, we propose an alternative method to produce independent samples from a conditioned distribution. The method learns the parameters of a generalized dynamical model that optimally describe the conditioned distribution in a variational sense. The outcome is an effective, unconditioned, dynamical model, from which one can trivially obtain independent samples, effectively restoring causality of the conditioned distribution. The consequences are twofold: on the one hand, it allows us to efficiently compute observables from the conditioned dynamics by simply averaging over independent samples. On the other hand, the method gives an effective unconditioned distribution which is easier to interpret. The method is flexible and can be applied virtually to any dynamics. We discuss an important application of the method, namely the problem of epidemic risk assessment from (imperfect) clinical tests, for a large family of time-continuous epidemic models endowed with a Gillespie-like sampler. We show that the method compares favorably against the state of the art, including the soft-margin approach and mean-field methods.
Sparse generative modeling of protein-sequence families
Nov 23, 2020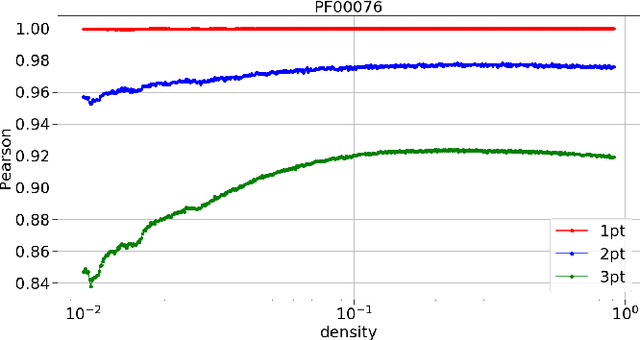
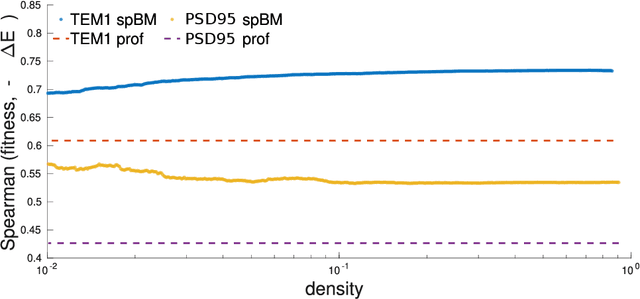
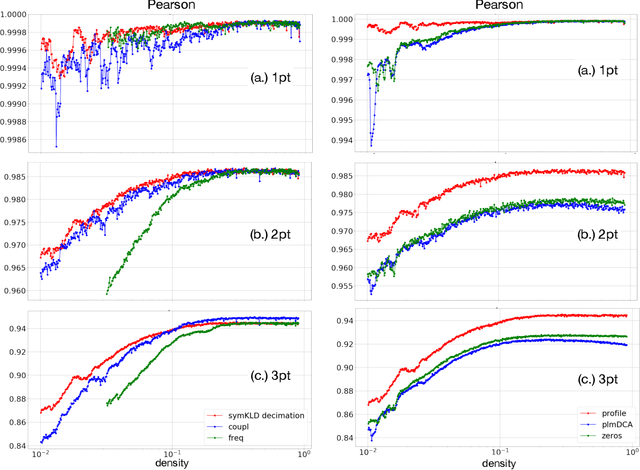
Pairwise Potts models (PM) provide accurate statistical models of families of evolutionarily related protein sequences. Their parameters are the local fields, which describe site-specific patterns of amino-acid conservation, and the two-site couplings, which mirror the coevolution between pairs of distinct sites. This coevolution reflects structural and functional constraints acting on protein sequences during evolution, and couplings can a priori connect any pairs of sites, even those being distant along the protein chain, or distant in the three-dimensional protein fold. The most conservative choice to describe all of the coevolution signal is to include all possible two-site couplings into the PM. This choice, typically made by what is known as Direct Coupling Analysis, has been highly successful in using sequences for predicting residue contacts in the three-dimensional structure, mutational effects, and in generating new functional sequences. However, the resulting PM suffers from important over-fitting effects: many couplings are small, noisy and hardly interpretable, and the PM is close to a critical point, meaning that it is highly sensitive to small parameter perturbations. In this work, we introduce a parameter-reduction procedure via iterative decimation of the less statistically significant couplings. We propose an information-based criterion that identifies couplings that are either weak, or statistically unsupported. We show that our procedure allows one to remove more than 90% of the PM couplings, while preserving the predictive and generative properties of the original dense PM. The resulting model is far away from criticality, meaning that it is more robust to noise, and its couplings are more easily interpretable.
Epidemic mitigation by statistical inference from contact tracing data
Sep 20, 2020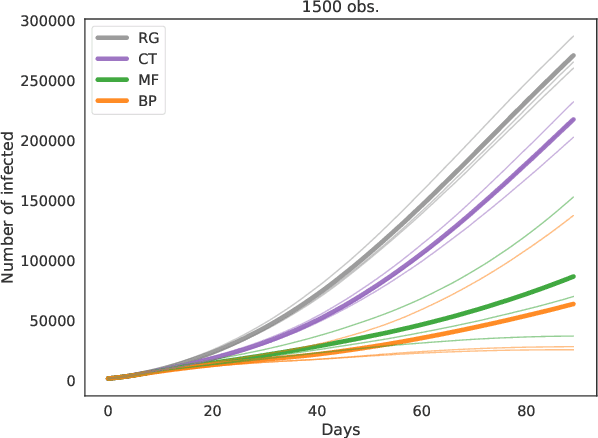
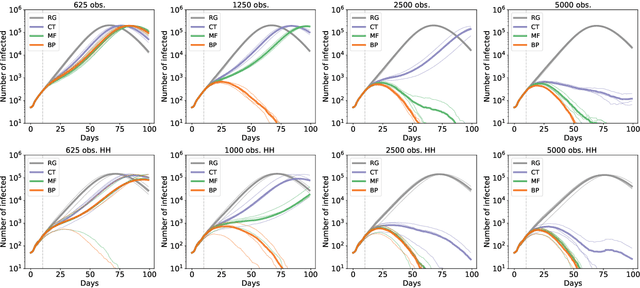
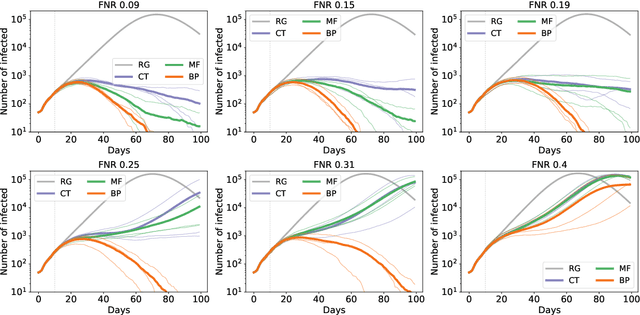
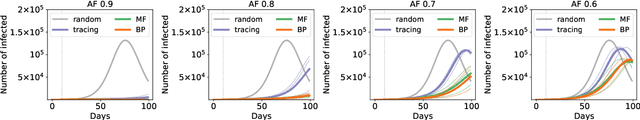
Contact-tracing is an essential tool in order to mitigate the impact of pandemic such as the COVID-19. In order to achieve efficient and scalable contact-tracing in real time, digital devices can play an important role. While a lot of attention has been paid to analyzing the privacy and ethical risks of the associated mobile applications, so far much less research has been devoted to optimizing their performance and assessing their impact on the mitigation of the epidemic. We develop Bayesian inference methods to estimate the risk that an individual is infected. This inference is based on the list of his recent contacts and their own risk levels, as well as personal information such as results of tests or presence of syndromes. We propose to use probabilistic risk estimation in order to optimize testing and quarantining strategies for the control of an epidemic. Our results show that in some range of epidemic spreading (typically when the manual tracing of all contacts of infected people becomes practically impossible, but before the fraction of infected people reaches the scale where a lock-down becomes unavoidable), this inference of individuals at risk could be an efficient way to mitigate the epidemic. Our approaches translate into fully distributed algorithms that only require communication between individuals who have recently been in contact. Such communication may be encrypted and anonymized and thus compatible with privacy preserving standards. We conclude that probabilistic risk estimation is capable to enhance performance of digital contact tracing and should be considered in the currently developed mobile applications.
Compressed sensing reconstruction using Expectation Propagation
Apr 10, 2019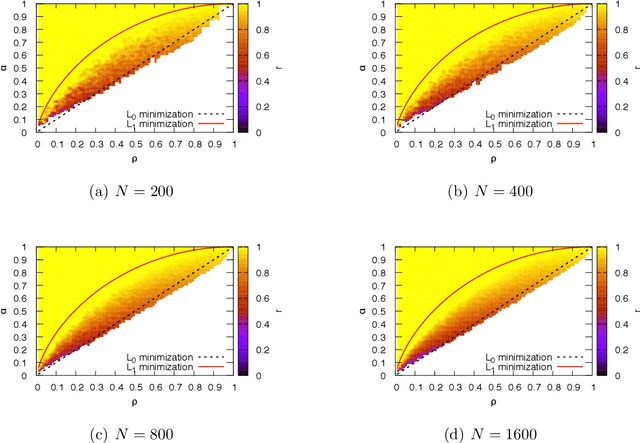
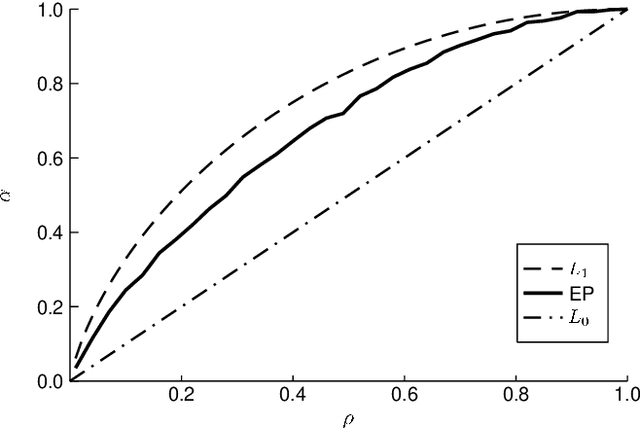
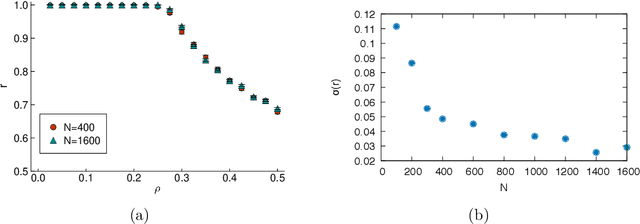
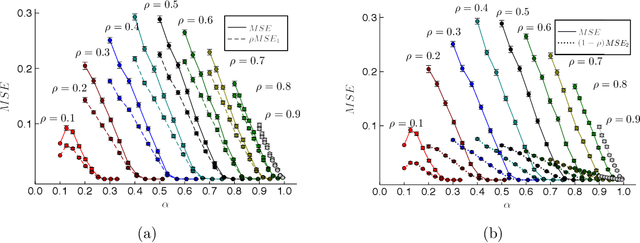
Many interesting problems in fields ranging from telecommunications to computational biology can be formalized in terms of large underdetermined systems of linear equations with additional constraints or regularizers. One of the most studied ones, the Compressed Sensing problem (CS), consists in finding the solution with the smallest number of non-zero components of a given system of linear equations $\boldsymbol y = \mathbf{F} \boldsymbol w$ for known measurement vector $\boldsymbol y$ and sensing matrix $\mathbf{F}$. Here, we will address the compressed sensing problem within a Bayesian inference framework where the sparsity constraint is remapped into a singular prior distribution (called Spike-and-Slab or Bernoulli-Gauss). Solution to the problem is attempted through the computation of marginal distributions via Expectation Propagation (EP), an iterative computational scheme originally developed in Statistical Physics. We will show that this strategy is comparatively more accurate than the alternatives in solving instances of CS generated from statistically correlated measurement matrices. For computational strategies based on the Bayesian framework such as variants of Belief Propagation, this is to be expected, as they implicitly rely on the hypothesis of statistical independence among the entries of the sensing matrix. Perhaps surprisingly, the method outperforms uniformly also all the other state-of-the-art methods in our tests.