 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeadabmDCA 2.0 -- a flexible but easy-to-use package for Direct Coupling Analysis
Jan 30, 2025In this methods article, we provide a flexible but easy-to-use implementation of Direct Coupling Analysis (DCA) based on Boltzmann machine learning, together with a tutorial on how to use it. The package \texttt{adabmDCA 2.0} is available in different programming languages (C++, Julia, Python) usable on different architectures (single-core and multi-core CPU, GPU) using a common front-end interface. In addition to several learning protocols for dense and sparse generative DCA models, it allows to directly address common downstream tasks like residue-residue contact prediction, mutational-effect prediction, scoring of sequence libraries and generation of artificial sequences for sequence design. It is readily applicable to protein and RNA sequence data.
Exact full-RSB SAT/UNSAT transition in infinitely wide two-layer neural networks
Oct 09, 2024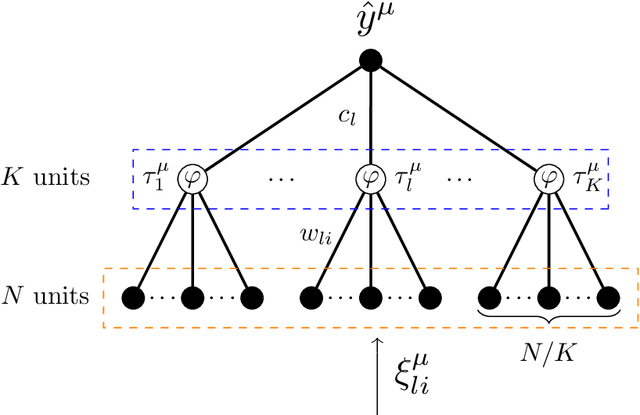
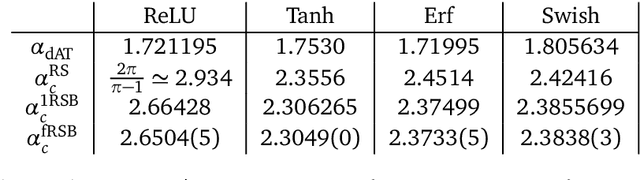
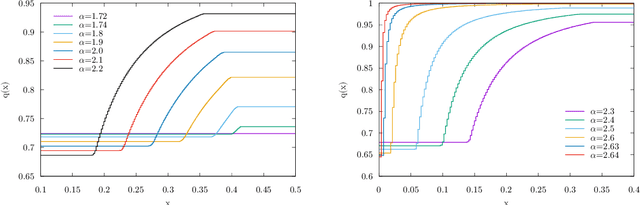
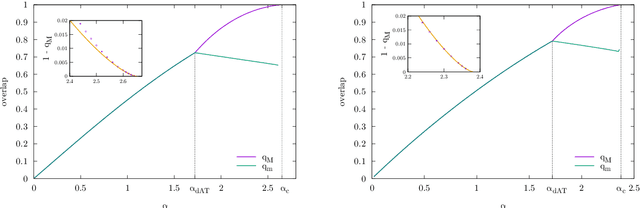
We analyze the problem of storing random pattern-label associations using two classes of continuous non-convex weights models, namely the perceptron with negative margin and an infinite width two layer neural network with non-overlapping receptive fields and generic activation function. Using a full-RSB ansatz we compute the exact value of the SAT/UNSAT transition. Furthermore, in the case of the negative perceptron model we show that, depending on the value of the margin and the constrained density, there is a line separating a phase in which the distribution of overlaps of typical states does not possess a gap from one in which it does. Our results show that the hypothesis underlying some recently developed theorems claiming that Approximate Message Passing (AMP) based algorithms are able to reach capacity, does not hold in general. Finally, we show that Gradient Descent is not able to reach the maximal capacity both in cases where there is and there is not a non-overlap gap phase for the typical states. This, similarly to what occurs in binary weight models, suggests that gradient-based algorithms are biased towards highly atypical states, whose inaccessibility determines the algorithmic threshold.
Sparse generative modeling of protein-sequence families
Nov 23, 2020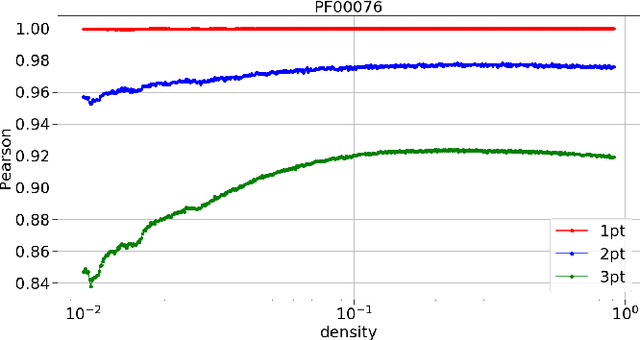
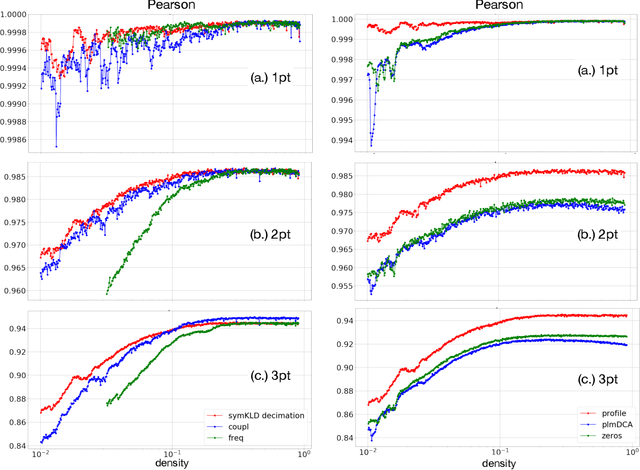
Pairwise Potts models (PM) provide accurate statistical models of families of evolutionarily related protein sequences. Their parameters are the local fields, which describe site-specific patterns of amino-acid conservation, and the two-site couplings, which mirror the coevolution between pairs of distinct sites. This coevolution reflects structural and functional constraints acting on protein sequences during evolution, and couplings can a priori connect any pairs of sites, even those being distant along the protein chain, or distant in the three-dimensional protein fold. The most conservative choice to describe all of the coevolution signal is to include all possible two-site couplings into the PM. This choice, typically made by what is known as Direct Coupling Analysis, has been highly successful in using sequences for predicting residue contacts in the three-dimensional structure, mutational effects, and in generating new functional sequences. However, the resulting PM suffers from important over-fitting effects: many couplings are small, noisy and hardly interpretable, and the PM is close to a critical point, meaning that it is highly sensitive to small parameter perturbations. In this work, we introduce a parameter-reduction procedure via iterative decimation of the less statistically significant couplings. We propose an information-based criterion that identifies couplings that are either weak, or statistically unsupported. We show that our procedure allows one to remove more than 90% of the PM couplings, while preserving the predictive and generative properties of the original dense PM. The resulting model is far away from criticality, meaning that it is more robust to noise, and its couplings are more easily interpretable.