 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeREN: Anatomically-Informed Mixture-of-Experts for Interstitial Lung Disease Diagnosis
Oct 06, 2025Mixture-of-Experts (MoE) architectures have significantly contributed to scalable machine learning by enabling specialized subnetworks to tackle complex tasks efficiently. However, traditional MoE systems lack domain-specific constraints essential for medical imaging, where anatomical structure and regional disease heterogeneity strongly influence pathological patterns. Here, we introduce Regional Expert Networks (REN), the first anatomically-informed MoE framework tailored specifically for medical image classification. REN leverages anatomical priors to train seven specialized experts, each dedicated to distinct lung lobes and bilateral lung combinations, enabling precise modeling of region-specific pathological variations. Multi-modal gating mechanisms dynamically integrate radiomics biomarkers and deep learning (DL) features (CNN, ViT, Mamba) to weight expert contributions optimally. Applied to interstitial lung disease (ILD) classification, REN achieves consistently superior performance: the radiomics-guided ensemble reached an average AUC of 0.8646 +/- 0.0467, a +12.5 percent improvement over the SwinUNETR baseline (AUC 0.7685, p = 0.031). Region-specific experts further revealed that lower-lobe models achieved AUCs of 0.88-0.90, surpassing DL counterparts (CNN: 0.76-0.79) and aligning with known disease progression patterns. Through rigorous patient-level cross-validation, REN demonstrates strong generalizability and clinical interpretability, presenting a scalable, anatomically-guided approach readily extensible to other structured medical imaging applications.
Towards Space Group Determination from EBSD Patterns: The Role of Deep Learning and High-throughput Dynamical Simulations
Apr 30, 2025The design of novel materials hinges on the understanding of structure-property relationships. However, our capability to synthesize a large number of materials has outpaced the ability and speed needed to characterize them. While the overall chemical constituents can be readily known during synthesis, the structural evolution and characterization of newly synthesized samples remains a bottleneck for the ultimate goal of high throughput nanomaterials discovery. Thus, scalable methods for crystal symmetry determination that can analyze a large volume of material samples within a short time-frame are especially needed. Kikuchi diffraction in the SEM is a promising technique for this due to its sensitivity to dynamical scattering, which may provide information beyond just the seven crystal systems and fourteen Bravais lattices. After diffraction patterns are collected from material samples, deep learning methods may be able to classify the space group symmetries using the patterns as input, which paired with the elemental composition, would help enable the determination of the crystal structure. To investigate the feasibility of this solution, neural networks were trained to predict the space group type of background corrected EBSD patterns. Our networks were first trained and tested on an artificial dataset of EBSD patterns of 5,148 different cubic phases, created through physics-based dynamical simulations. Next, Maximum Classifier Discrepancy, an unsupervised deep learning-based domain adaptation method, was utilized to train neural networks to make predictions for experimental EBSD patterns. We introduce a relabeling scheme, which enables our models to achieve accuracy scores higher than 90% on simulated and experimental data, suggesting that neural networks are capable of making predictions of crystal symmetry from an EBSD pattern.
An Incremental Phase Mapping Approach for X-ray Diffraction Patterns using Binary Peak Representations
Nov 08, 2022



Despite the huge advancement in knowledge discovery and data mining techniques, the X-ray diffraction (XRD) analysis process has mostly remained untouched and still involves manual investigation, comparison, and verification. Due to the large volume of XRD samples from high-throughput XRD experiments, it has become impossible for domain scientists to process them manually. Recently, they have started leveraging standard clustering techniques, to reduce the XRD pattern representations requiring manual efforts for labeling and verification. Nevertheless, these standard clustering techniques do not handle problem-specific aspects such as peak shifting, adjacent peaks, background noise, and mixed phases; hence, resulting in incorrect composition-phase diagrams that complicate further steps. Here, we leverage data mining techniques along with domain expertise to handle these issues. In this paper, we introduce an incremental phase mapping approach based on binary peak representations using a new threshold based fuzzy dissimilarity measure. The proposed approach first applies an incremental phase computation algorithm on discrete binary peak representation of XRD samples, followed by hierarchical clustering or manual merging of similar pure phases to obtain the final composition-phase diagram. We evaluate our method on the composition space of two ternary alloy systems- Co-Ni-Ta and Co-Ti-Ta. Our results are verified by domain scientists and closely resembles the manually computed ground-truth composition-phase diagrams. The proposed approach takes us closer towards achieving the goal of complete end-to-end automated XRD analysis.
A General Framework Combining Generative Adversarial Networks and Mixture Density Networks for Inverse Modeling in Microstructural Materials Design
Jan 26, 2021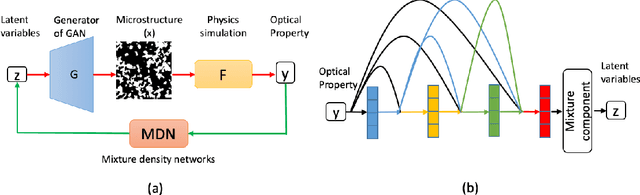



Microstructural materials design is one of the most important applications of inverse modeling in materials science. Generally speaking, there are two broad modeling paradigms in scientific applications: forward and inverse. While the forward modeling estimates the observations based on known parameters, the inverse modeling attempts to infer the parameters given the observations. Inverse problems are usually more critical as well as difficult in scientific applications as they seek to explore the parameters that cannot be directly observed. Inverse problems are used extensively in various scientific fields, such as geophysics, healthcare and materials science. However, it is challenging to solve inverse problems, because they usually need to learn a one-to-many non-linear mapping, and also require significant computing time, especially for high-dimensional parameter space. Further, inverse problems become even more difficult to solve when the dimension of input (i.e. observation) is much lower than that of output (i.e. parameters). In this work, we propose a framework consisting of generative adversarial networks and mixture density networks for inverse modeling, and it is evaluated on a materials science dataset for microstructural materials design. Compared with baseline methods, the results demonstrate that the proposed framework can overcome the above-mentioned challenges and produce multiple promising solutions in an efficient manner.
A real-time iterative machine learning approach for temperature profile prediction in additive manufacturing processes
Aug 09, 2019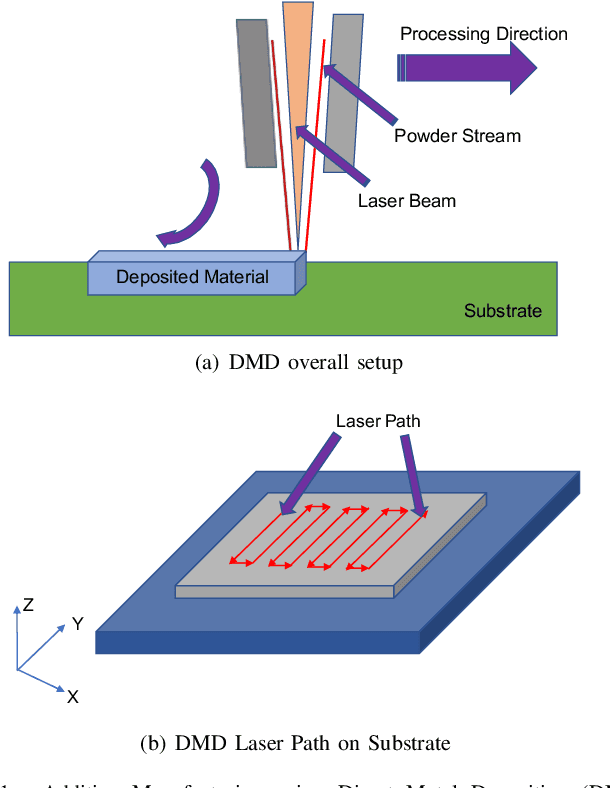

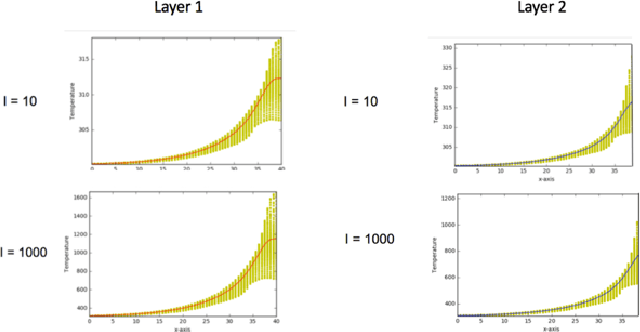
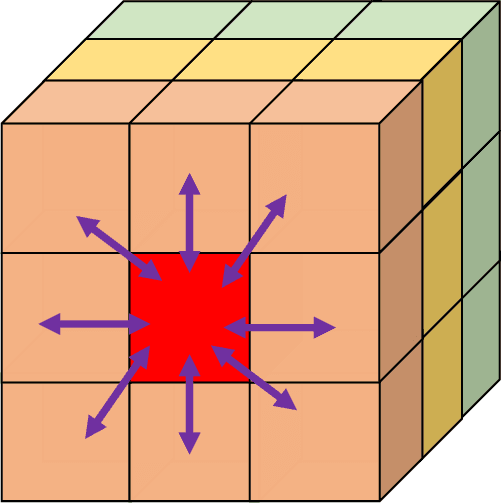
Additive Manufacturing (AM) is a manufacturing paradigm that builds three-dimensional objects from a computer-aided design model by successively adding material layer by layer. AM has become very popular in the past decade due to its utility for fast prototyping such as 3D printing as well as manufacturing functional parts with complex geometries using processes such as laser metal deposition that would be difficult to create using traditional machining. As the process for creating an intricate part for an expensive metal such as Titanium is prohibitive with respect to cost, computational models are used to simulate the behavior of AM processes before the experimental run. However, as the simulations are computationally costly and time-consuming for predicting multiscale multi-physics phenomena in AM, physics-informed data-driven machine-learning systems for predicting the behavior of AM processes are immensely beneficial. Such models accelerate not only multiscale simulation tools but also empower real-time control systems using in-situ data. In this paper, we design and develop essential components of a scientific framework for developing a data-driven model-based real-time control system. Finite element methods are employed for solving time-dependent heat equations and developing the database. The proposed framework uses extremely randomized trees - an ensemble of bagged decision trees as the regression algorithm iteratively using temperatures of prior voxels and laser information as inputs to predict temperatures of subsequent voxels. The models achieve mean absolute percentage errors below 1% for predicting temperature profiles for AM processes. The code is made available for the research community at https://github.com/paularindam/ml-iter-additive.
* 10 pages, 8 figures
IRNet: A General Purpose Deep Residual Regression Framework for Materials Discovery
Jul 07, 2019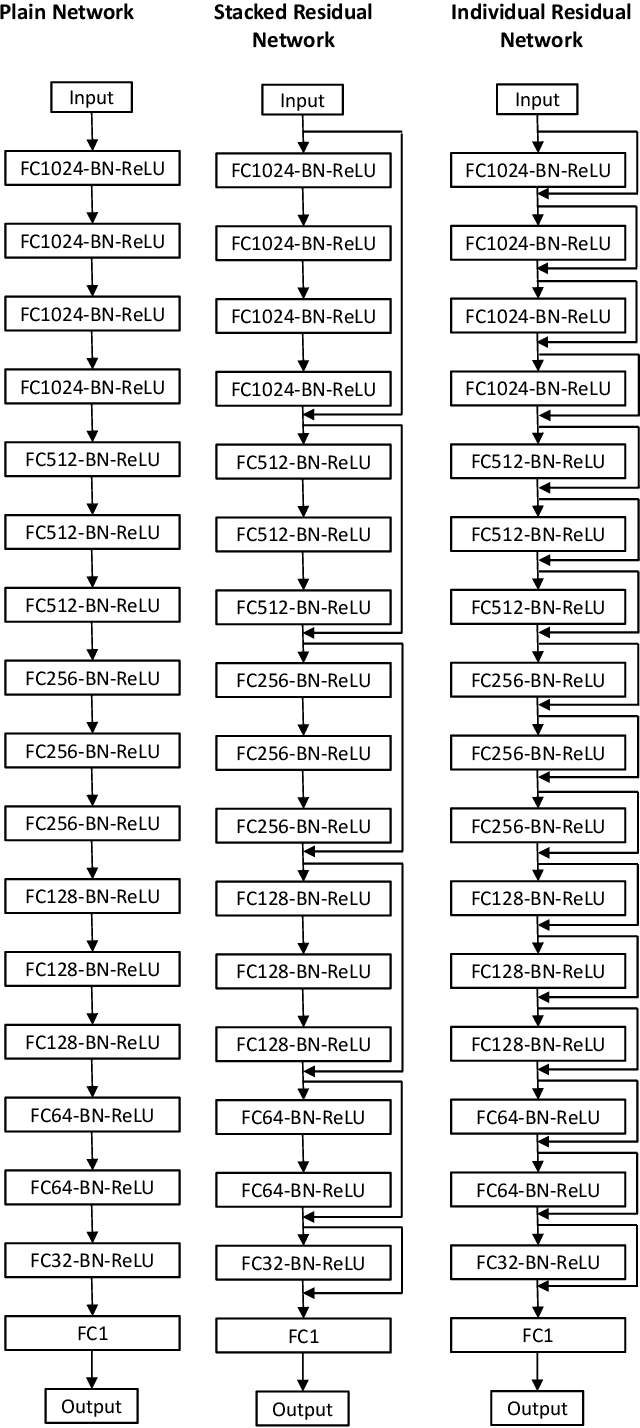



Materials discovery is crucial for making scientific advances in many domains. Collections of data from experiments and first-principle computations have spurred interest in applying machine learning methods to create predictive models capable of mapping from composition and crystal structures to materials properties. Generally, these are regression problems with the input being a 1D vector composed of numerical attributes representing the material composition and/or crystal structure. While neural networks consisting of fully connected layers have been applied to such problems, their performance often suffers from the vanishing gradient problem when network depth is increased. In this paper, we study and propose design principles for building deep regression networks composed of fully connected layers with numerical vectors as input. We introduce a novel deep regression network with individual residual learning, IRNet, that places shortcut connections after each layer so that each layer learns the residual mapping between its output and input. We use the problem of learning properties of inorganic materials from numerical attributes derived from material composition and/or crystal structure to compare IRNet's performance against that of other machine learning techniques. Using multiple datasets from the Open Quantum Materials Database (OQMD) and Materials Project for training and evaluation, we show that IRNet provides significantly better prediction performance than the state-of-the-art machine learning approaches currently used by domain scientists. We also show that IRNet's use of individual residual learning leads to better convergence during the training phase than when shortcut connections are between multi-layer stacks while maintaining the same number of parameters.
Transfer Learning Using Ensemble Neural Networks for Organic Solar Cell Screening
Mar 30, 2019



Organic Solar Cells are a promising technology for solving the clean energy crisis in the world. However, generating candidate chemical compounds for solar cells is a time-consuming process requiring thousands of hours of laboratory analysis. For a solar cell, the most important property is the power conversion efficiency which is dependent on the highest occupied molecular orbitals (HOMO) values of the donor molecules. Recently, machine learning techniques have proved to be very useful in building predictive models for HOMO values of donor structures of Organic Photovoltaic Cells (OPVs). Since experimental datasets are limited in size, current machine learning models are trained on data derived from calculations based on density functional theory (DFT). Molecular line notations such as SMILES or InChI are popular input representations for describing the molecular structure of donor molecules. The two types of line representations encode different information, such as SMILES defines the bond types while InChi defines protonation. In this work, we present an ensemble deep neural network architecture, called SINet, which harnesses both the SMILES and InChI molecular representations to predict HOMO values and leverage the potential of transfer learning from a sizeable DFT-computed dataset- Harvard CEP to build more robust predictive models for relatively smaller HOPV datasets. Harvard CEP dataset contains molecular structures and properties for 2.3 million candidate donor structures for OPV while HOPV contains DFT-computed and experimental values of 350 and 243 molecules respectively. Our results demonstrate significant performance improvement from the use of transfer learning and leveraging both molecular representations.
CheMixNet: Mixed DNN Architectures for Predicting Chemical Properties using Multiple Molecular Representations
Nov 30, 2018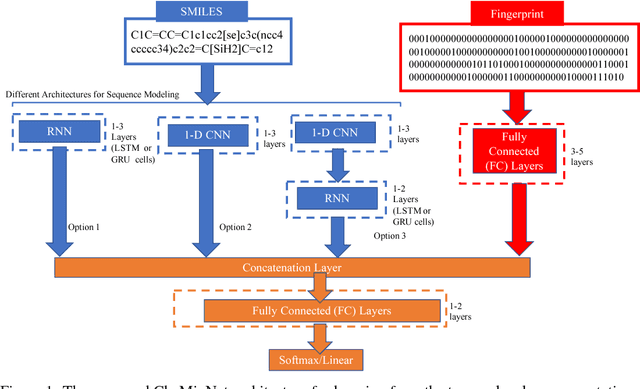
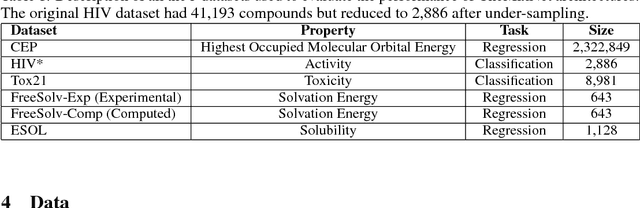
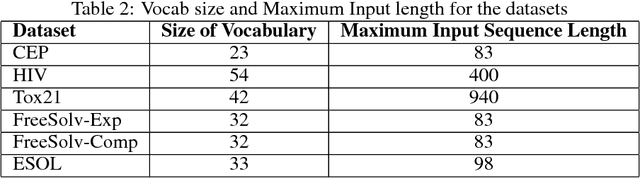

SMILES is a linear representation of chemical structures which encodes the connection table, and the stereochemistry of a molecule as a line of text with a grammar structure denoting atoms, bonds, rings and chains, and this information can be used to predict chemical properties. Molecular fingerprints are representations of chemical structures, successfully used in similarity search, clustering, classification, drug discovery, and virtual screening and are a standard and computationally efficient abstract representation where structural features are represented as a bit string. Both SMILES and molecular fingerprints are different representations for describing the structure of a molecule. There exist several predictive models for learning chemical properties based on either SMILES or molecular fingerprints. Here, our goal is to build predictive models that can leverage both these molecular representations. In this work, we present CheMixNet -- a set of neural networks for predicting chemical properties from a mixture of features learned from the two molecular representations -- SMILES as sequences and molecular fingerprints as vector inputs. We demonstrate the efficacy of CheMixNet architectures by evaluating on six different datasets. The proposed CheMixNet models not only outperforms the candidate neural architectures such as contemporary fully connected networks that uses molecular fingerprints and 1-D CNN and RNN models trained SMILES sequences, but also other state-of-the-art architectures such as Chemception and Molecular Graph Convolutions.
A New Parallel Algorithm for Two-Pass Connected Component Labeling
Jun 20, 2016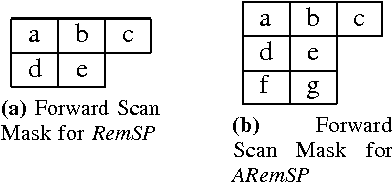
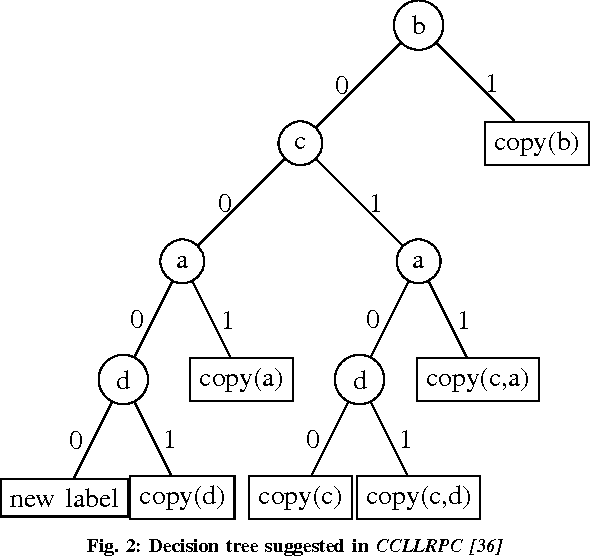
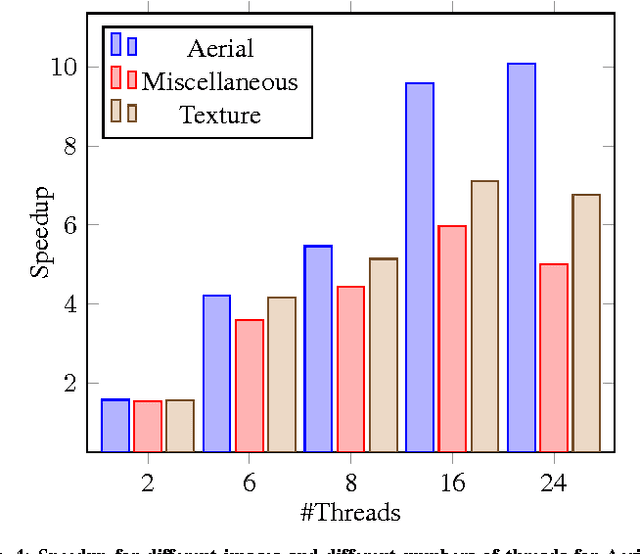
Connected Component Labeling (CCL) is an important step in pattern recognition and image processing. It assigns labels to the pixels such that adjacent pixels sharing the same features are assigned the same label. Typically, CCL requires several passes over the data. We focus on two-pass technique where each pixel is given a provisional label in the first pass whereas an actual label is assigned in the second pass. We present a scalable parallel two-pass CCL algorithm, called PAREMSP, which employs a scan strategy and the best union-find technique called REMSP, which uses REM's algorithm for storing label equivalence information of pixels in a 2-D image. In the first pass, we divide the image among threads and each thread runs the scan phase along with REMSP simultaneously. In the second phase, we assign the final labels to the pixels. As REMSP is easily parallelizable, we use the parallel version of REMSP for merging the pixels on the boundary. Our experiments show the scalability of PAREMSP achieving speedups up to $20.1$ using $24$ cores on shared memory architecture using OpenMP for an image of size $465.20$ MB. We find that our proposed parallel algorithm achieves linear scaling for a large resolution fixed problem size as the number of processing elements are increased. Additionally, the parallel algorithm does not make use of any hardware specific routines, and thus is highly portable.
An exploration of parameter redundancy in deep networks with circulant projections
Oct 27, 2015
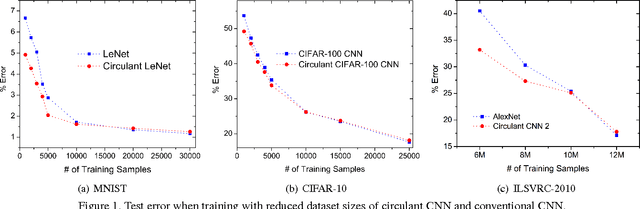
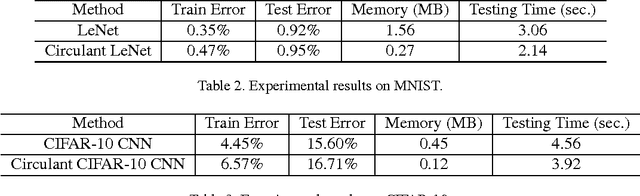

We explore the redundancy of parameters in deep neural networks by replacing the conventional linear projection in fully-connected layers with the circulant projection. The circulant structure substantially reduces memory footprint and enables the use of the Fast Fourier Transform to speed up the computation. Considering a fully-connected neural network layer with d input nodes, and d output nodes, this method improves the time complexity from O(d^2) to O(dlogd) and space complexity from O(d^2) to O(d). The space savings are particularly important for modern deep convolutional neural network architectures, where fully-connected layers typically contain more than 90% of the network parameters. We further show that the gradient computation and optimization of the circulant projections can be performed very efficiently. Our experiments on three standard datasets show that the proposed approach achieves this significant gain in storage and efficiency with minimal increase in error rate compared to neural networks with unstructured projections.