 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTransfer Learning Using Ensemble Neural Networks for Organic Solar Cell Screening
Mar 30, 2019



Organic Solar Cells are a promising technology for solving the clean energy crisis in the world. However, generating candidate chemical compounds for solar cells is a time-consuming process requiring thousands of hours of laboratory analysis. For a solar cell, the most important property is the power conversion efficiency which is dependent on the highest occupied molecular orbitals (HOMO) values of the donor molecules. Recently, machine learning techniques have proved to be very useful in building predictive models for HOMO values of donor structures of Organic Photovoltaic Cells (OPVs). Since experimental datasets are limited in size, current machine learning models are trained on data derived from calculations based on density functional theory (DFT). Molecular line notations such as SMILES or InChI are popular input representations for describing the molecular structure of donor molecules. The two types of line representations encode different information, such as SMILES defines the bond types while InChi defines protonation. In this work, we present an ensemble deep neural network architecture, called SINet, which harnesses both the SMILES and InChI molecular representations to predict HOMO values and leverage the potential of transfer learning from a sizeable DFT-computed dataset- Harvard CEP to build more robust predictive models for relatively smaller HOPV datasets. Harvard CEP dataset contains molecular structures and properties for 2.3 million candidate donor structures for OPV while HOPV contains DFT-computed and experimental values of 350 and 243 molecules respectively. Our results demonstrate significant performance improvement from the use of transfer learning and leveraging both molecular representations.
CheMixNet: Mixed DNN Architectures for Predicting Chemical Properties using Multiple Molecular Representations
Nov 30, 2018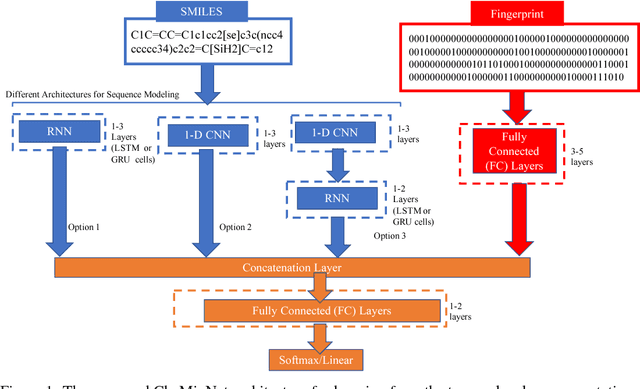
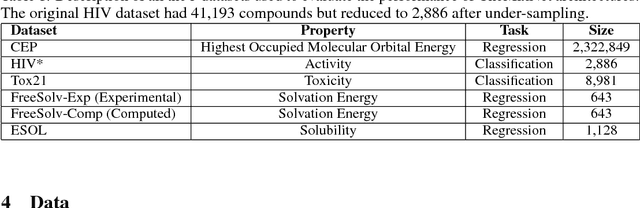
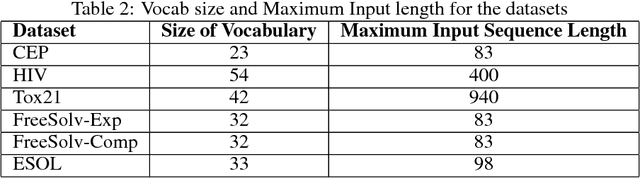

SMILES is a linear representation of chemical structures which encodes the connection table, and the stereochemistry of a molecule as a line of text with a grammar structure denoting atoms, bonds, rings and chains, and this information can be used to predict chemical properties. Molecular fingerprints are representations of chemical structures, successfully used in similarity search, clustering, classification, drug discovery, and virtual screening and are a standard and computationally efficient abstract representation where structural features are represented as a bit string. Both SMILES and molecular fingerprints are different representations for describing the structure of a molecule. There exist several predictive models for learning chemical properties based on either SMILES or molecular fingerprints. Here, our goal is to build predictive models that can leverage both these molecular representations. In this work, we present CheMixNet -- a set of neural networks for predicting chemical properties from a mixture of features learned from the two molecular representations -- SMILES as sequences and molecular fingerprints as vector inputs. We demonstrate the efficacy of CheMixNet architectures by evaluating on six different datasets. The proposed CheMixNet models not only outperforms the candidate neural architectures such as contemporary fully connected networks that uses molecular fingerprints and 1-D CNN and RNN models trained SMILES sequences, but also other state-of-the-art architectures such as Chemception and Molecular Graph Convolutions.