 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAn Incremental Phase Mapping Approach for X-ray Diffraction Patterns using Binary Peak Representations
Nov 08, 2022



Despite the huge advancement in knowledge discovery and data mining techniques, the X-ray diffraction (XRD) analysis process has mostly remained untouched and still involves manual investigation, comparison, and verification. Due to the large volume of XRD samples from high-throughput XRD experiments, it has become impossible for domain scientists to process them manually. Recently, they have started leveraging standard clustering techniques, to reduce the XRD pattern representations requiring manual efforts for labeling and verification. Nevertheless, these standard clustering techniques do not handle problem-specific aspects such as peak shifting, adjacent peaks, background noise, and mixed phases; hence, resulting in incorrect composition-phase diagrams that complicate further steps. Here, we leverage data mining techniques along with domain expertise to handle these issues. In this paper, we introduce an incremental phase mapping approach based on binary peak representations using a new threshold based fuzzy dissimilarity measure. The proposed approach first applies an incremental phase computation algorithm on discrete binary peak representation of XRD samples, followed by hierarchical clustering or manual merging of similar pure phases to obtain the final composition-phase diagram. We evaluate our method on the composition space of two ternary alloy systems- Co-Ni-Ta and Co-Ti-Ta. Our results are verified by domain scientists and closely resembles the manually computed ground-truth composition-phase diagrams. The proposed approach takes us closer towards achieving the goal of complete end-to-end automated XRD analysis.
Designing an Efficient End-to-end Machine Learning Pipeline for Real-time Empty-shelf Detection
May 28, 2022
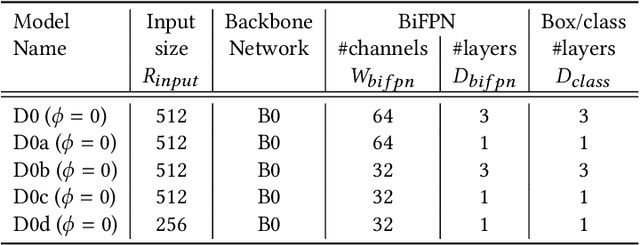
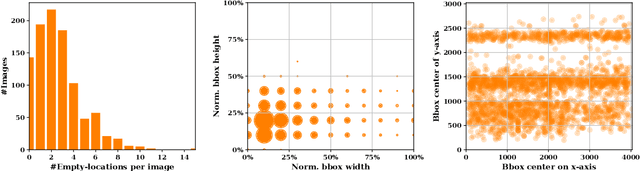
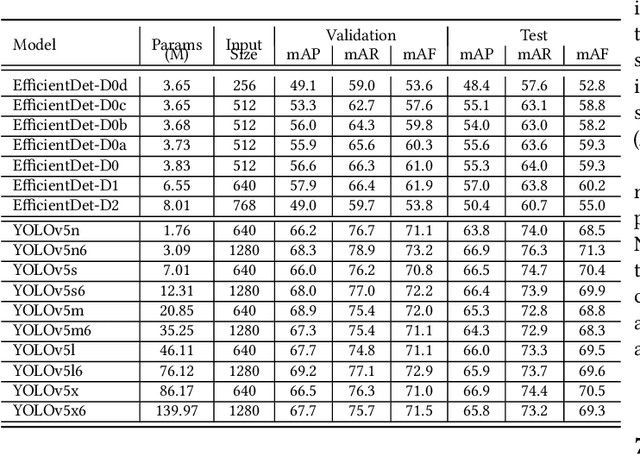
On-Shelf Availability (OSA) of products in retail stores is a critical business criterion in the fast moving consumer goods and retails sector. When a product is out-of-stock (OOS) and a customer cannot find it on its designed shelf, this motivates the customer to store-switching or buying nothing, which causes fall in future sales and demands. Retailers are employing several approaches to detect empty shelves and ensure high OSA of products; however, such methods are generally ineffective and infeasible since they are either manual, expensive or less accurate. Recently machine learning based solutions have been proposed, but they suffer from high computational cost and low accuracy problem due to lack of large annotated datasets of on-shelf products. Here, we present an elegant approach for designing an end-to-end machine learning (ML) pipeline for real-time empty shelf detection. Considering the strong dependency between the quality of ML models and the quality of data, we focus on the importance of proper data collection, cleaning and correct data annotation before delving into modeling. Since an empty-shelf detection solution should be computationally-efficient for real-time predictions, we explore different run-time optimizations to improve the model performance. Our dataset contains 1000 images, collected and annotated by following well-defined guidelines. Our low-latency model achieves a mean average F1-score of 68.5%, and can process up to 67 images/s on Intel Xeon Gold and up to 860 images/s on an A100 GPU.
A General Framework Combining Generative Adversarial Networks and Mixture Density Networks for Inverse Modeling in Microstructural Materials Design
Jan 26, 2021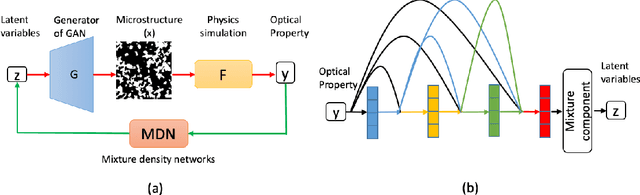



Microstructural materials design is one of the most important applications of inverse modeling in materials science. Generally speaking, there are two broad modeling paradigms in scientific applications: forward and inverse. While the forward modeling estimates the observations based on known parameters, the inverse modeling attempts to infer the parameters given the observations. Inverse problems are usually more critical as well as difficult in scientific applications as they seek to explore the parameters that cannot be directly observed. Inverse problems are used extensively in various scientific fields, such as geophysics, healthcare and materials science. However, it is challenging to solve inverse problems, because they usually need to learn a one-to-many non-linear mapping, and also require significant computing time, especially for high-dimensional parameter space. Further, inverse problems become even more difficult to solve when the dimension of input (i.e. observation) is much lower than that of output (i.e. parameters). In this work, we propose a framework consisting of generative adversarial networks and mixture density networks for inverse modeling, and it is evaluated on a materials science dataset for microstructural materials design. Compared with baseline methods, the results demonstrate that the proposed framework can overcome the above-mentioned challenges and produce multiple promising solutions in an efficient manner.
IRNet: A General Purpose Deep Residual Regression Framework for Materials Discovery
Jul 07, 2019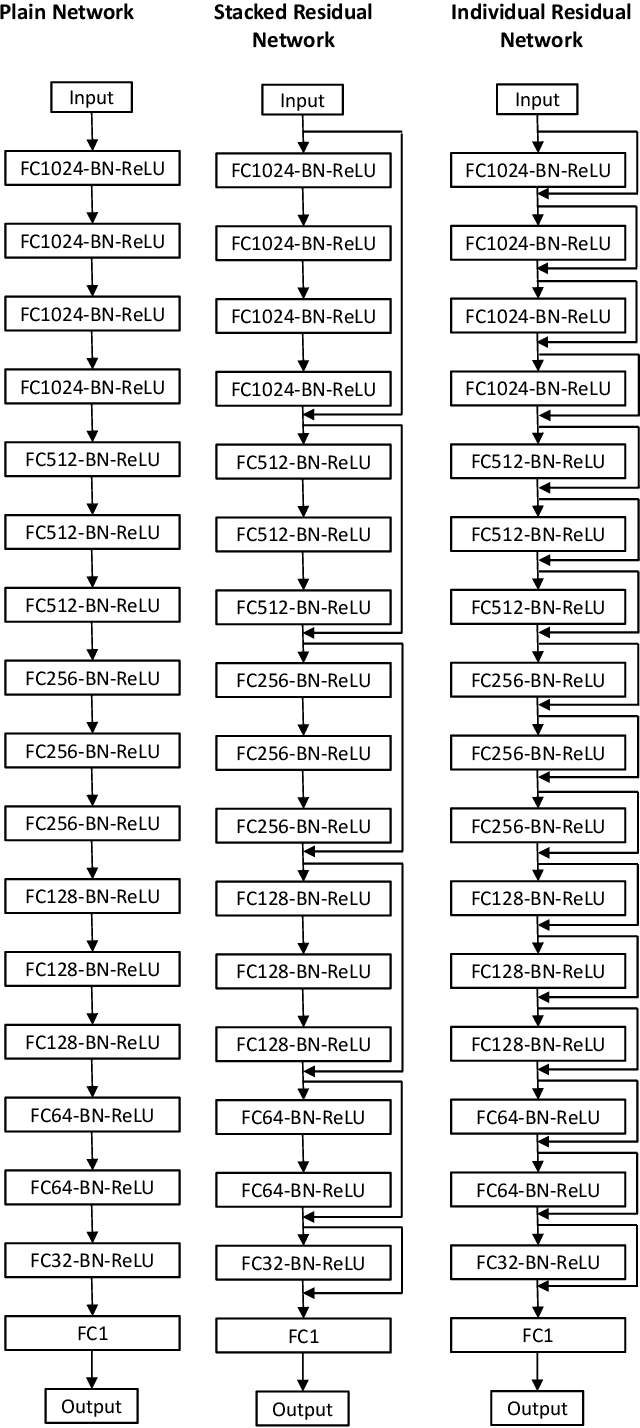



Materials discovery is crucial for making scientific advances in many domains. Collections of data from experiments and first-principle computations have spurred interest in applying machine learning methods to create predictive models capable of mapping from composition and crystal structures to materials properties. Generally, these are regression problems with the input being a 1D vector composed of numerical attributes representing the material composition and/or crystal structure. While neural networks consisting of fully connected layers have been applied to such problems, their performance often suffers from the vanishing gradient problem when network depth is increased. In this paper, we study and propose design principles for building deep regression networks composed of fully connected layers with numerical vectors as input. We introduce a novel deep regression network with individual residual learning, IRNet, that places shortcut connections after each layer so that each layer learns the residual mapping between its output and input. We use the problem of learning properties of inorganic materials from numerical attributes derived from material composition and/or crystal structure to compare IRNet's performance against that of other machine learning techniques. Using multiple datasets from the Open Quantum Materials Database (OQMD) and Materials Project for training and evaluation, we show that IRNet provides significantly better prediction performance than the state-of-the-art machine learning approaches currently used by domain scientists. We also show that IRNet's use of individual residual learning leads to better convergence during the training phase than when shortcut connections are between multi-layer stacks while maintaining the same number of parameters.
Transfer Learning Using Ensemble Neural Networks for Organic Solar Cell Screening
Mar 30, 2019



Organic Solar Cells are a promising technology for solving the clean energy crisis in the world. However, generating candidate chemical compounds for solar cells is a time-consuming process requiring thousands of hours of laboratory analysis. For a solar cell, the most important property is the power conversion efficiency which is dependent on the highest occupied molecular orbitals (HOMO) values of the donor molecules. Recently, machine learning techniques have proved to be very useful in building predictive models for HOMO values of donor structures of Organic Photovoltaic Cells (OPVs). Since experimental datasets are limited in size, current machine learning models are trained on data derived from calculations based on density functional theory (DFT). Molecular line notations such as SMILES or InChI are popular input representations for describing the molecular structure of donor molecules. The two types of line representations encode different information, such as SMILES defines the bond types while InChi defines protonation. In this work, we present an ensemble deep neural network architecture, called SINet, which harnesses both the SMILES and InChI molecular representations to predict HOMO values and leverage the potential of transfer learning from a sizeable DFT-computed dataset- Harvard CEP to build more robust predictive models for relatively smaller HOPV datasets. Harvard CEP dataset contains molecular structures and properties for 2.3 million candidate donor structures for OPV while HOPV contains DFT-computed and experimental values of 350 and 243 molecules respectively. Our results demonstrate significant performance improvement from the use of transfer learning and leveraging both molecular representations.
CheMixNet: Mixed DNN Architectures for Predicting Chemical Properties using Multiple Molecular Representations
Nov 30, 2018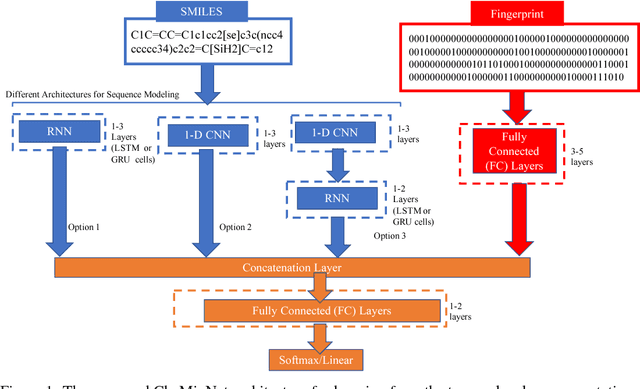
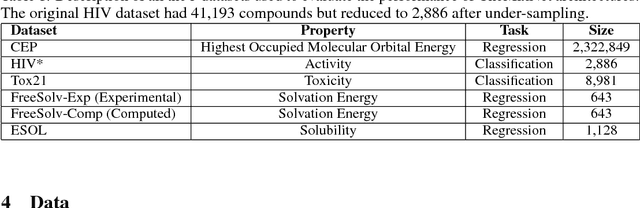
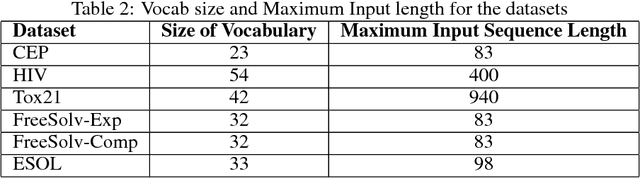

SMILES is a linear representation of chemical structures which encodes the connection table, and the stereochemistry of a molecule as a line of text with a grammar structure denoting atoms, bonds, rings and chains, and this information can be used to predict chemical properties. Molecular fingerprints are representations of chemical structures, successfully used in similarity search, clustering, classification, drug discovery, and virtual screening and are a standard and computationally efficient abstract representation where structural features are represented as a bit string. Both SMILES and molecular fingerprints are different representations for describing the structure of a molecule. There exist several predictive models for learning chemical properties based on either SMILES or molecular fingerprints. Here, our goal is to build predictive models that can leverage both these molecular representations. In this work, we present CheMixNet -- a set of neural networks for predicting chemical properties from a mixture of features learned from the two molecular representations -- SMILES as sequences and molecular fingerprints as vector inputs. We demonstrate the efficacy of CheMixNet architectures by evaluating on six different datasets. The proposed CheMixNet models not only outperforms the candidate neural architectures such as contemporary fully connected networks that uses molecular fingerprints and 1-D CNN and RNN models trained SMILES sequences, but also other state-of-the-art architectures such as Chemception and Molecular Graph Convolutions.