 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCardinality-Preserving Structured Sparse Graph Transformers for Molecular Property Prediction
Feb 02, 2026Drug discovery motivates efficient molecular property prediction under limited labeled data. Chemical space is vast, often estimated at approximately 10^60 drug-like molecules, while only thousands of drugs have been approved. As a result, self-supervised pretraining on large unlabeled molecular corpora has become essential for data-efficient molecular representation learning. We introduce **CardinalGraphFormer**, a graph transformer that incorporates Graphormer-inspired structural biases, including shortest-path distance and centrality, as well as direct-bond edge bias, within a structured sparse attention regime limited to shortest-path distance <= 3. The model further augments this design with a cardinality-preserving unnormalized aggregation channel over the same support set. Pretraining combines contrastive graph-level alignment with masked attribute reconstruction. Under a fully matched evaluation protocol, CardinalGraphFormer improves mean performance across all 11 evaluated tasks and achieves statistically significant gains on 10 of 11 public benchmarks spanning MoleculeNet, OGB, and TDC ADMET tasks when compared to strong reproduced baselines.
Decoding Futures Price Dynamics: A Regularized Sparse Autoencoder for Interpretable Multi-Horizon Forecasting and Factor Discovery
May 14, 2025Commodity price volatility creates economic challenges, necessitating accurate multi-horizon forecasting. Predicting prices for commodities like copper and crude oil is complicated by diverse interacting factors (macroeconomic, supply/demand, geopolitical, etc.). Current models often lack transparency, limiting strategic use. This paper presents a Regularized Sparse Autoencoder (RSAE), a deep learning framework for simultaneous multi-horizon commodity price prediction and discovery of interpretable latent market drivers. The RSAE forecasts prices at multiple horizons (e.g., 1-day, 1-week, 1-month) using multivariate time series. Crucially, L1 regularization ($\|\mathbf{z}\|_1$) on its latent vector $\mathbf{z}$ enforces sparsity, promoting parsimonious explanations of market dynamics through learned factors representing underlying drivers (e.g., demand, supply shocks). Drawing from energy-based models and sparse coding, the RSAE optimizes predictive accuracy while learning sparse representations. Evaluated on historical Copper and Crude Oil data with numerous indicators, our findings indicate the RSAE offers competitive multi-horizon forecasting accuracy and data-driven insights into price dynamics via its interpretable latent space, a key advantage over traditional black-box approaches.
D4Explainer: In-Distribution GNN Explanations via Discrete Denoising Diffusion
Oct 30, 2023The widespread deployment of Graph Neural Networks (GNNs) sparks significant interest in their explainability, which plays a vital role in model auditing and ensuring trustworthy graph learning. The objective of GNN explainability is to discern the underlying graph structures that have the most significant impact on model predictions. Ensuring that explanations generated are reliable necessitates consideration of the in-distribution property, particularly due to the vulnerability of GNNs to out-of-distribution data. Unfortunately, prevailing explainability methods tend to constrain the generated explanations to the structure of the original graph, thereby downplaying the significance of the in-distribution property and resulting in explanations that lack reliability. To address these challenges, we propose D4Explainer, a novel approach that provides in-distribution GNN explanations for both counterfactual and model-level explanation scenarios. The proposed D4Explainer incorporates generative graph distribution learning into the optimization objective, which accomplishes two goals: 1) generate a collection of diverse counterfactual graphs that conform to the in-distribution property for a given instance, and 2) identify the most discriminative graph patterns that contribute to a specific class prediction, thus serving as model-level explanations. It is worth mentioning that D4Explainer is the first unified framework that combines both counterfactual and model-level explanations. Empirical evaluations conducted on synthetic and real-world datasets provide compelling evidence of the state-of-the-art performance achieved by D4Explainer in terms of explanation accuracy, faithfulness, diversity, and robustness.
CardiGraphormer: Unveiling the Power of Self-Supervised Learning in Revolutionizing Drug Discovery
Jul 04, 2023In the expansive realm of drug discovery, with approximately 15,000 known drugs and only around 4,200 approved, the combinatorial nature of the chemical space presents a formidable challenge. While Artificial Intelligence (AI) has emerged as a powerful ally, traditional AI frameworks face significant hurdles. This manuscript introduces CardiGraphormer, a groundbreaking approach that synergizes self-supervised learning (SSL), Graph Neural Networks (GNNs), and Cardinality Preserving Attention to revolutionize drug discovery. CardiGraphormer, a novel combination of Graphormer and Cardinality Preserving Attention, leverages SSL to learn potent molecular representations and employs GNNs to extract molecular fingerprints, enhancing predictive performance and interpretability while reducing computation time. It excels in handling complex data like molecular structures and performs tasks associated with nodes, pairs of nodes, subgraphs, or entire graph structures. CardiGraphormer's potential applications in drug discovery and drug interactions are vast, from identifying new drug targets to predicting drug-to-drug interactions and enabling novel drug discovery. This innovative approach provides an AI-enhanced methodology in drug development, utilizing SSL combined with GNNs to overcome existing limitations and pave the way for a richer exploration of the vast combinatorial chemical space in drug discovery.
Prediction of good reaction coordinates and future evolution of MD trajectories using Regularized Sparse Autoencoders: A novel deep learning approach
Aug 22, 2022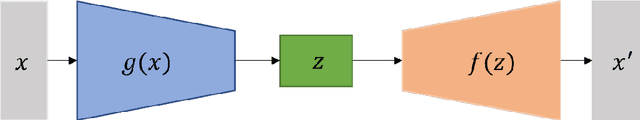

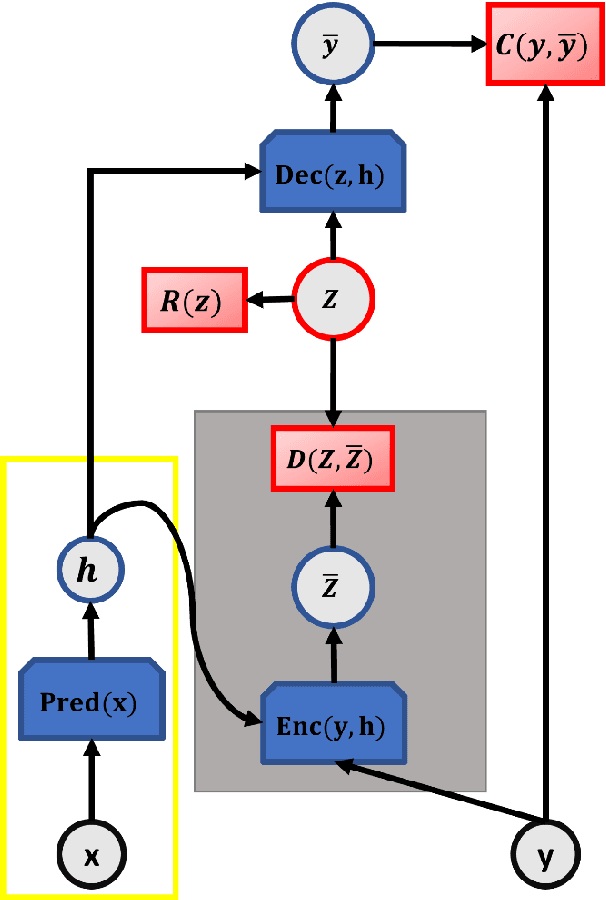
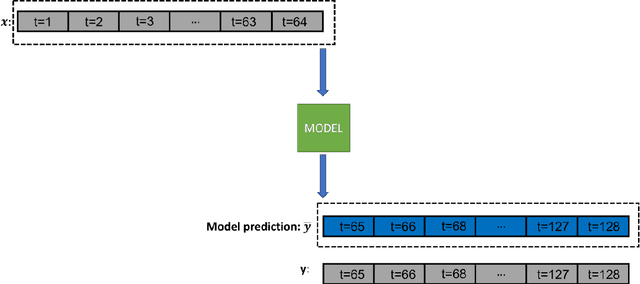
Identifying reaction coordinates(RCs) is an active area of research, given the crucial role RCs play in determining the progress of a chemical reaction. The choice of the reaction coordinate is often based on heuristic knowledge. However, an essential criterion for the choice is that the coordinate should capture both the reactant and product states unequivocally. Also, the coordinate should be the slowest one so that all the other degrees of freedom can easily equilibrate along the reaction coordinate. Also, the coordinate should be the slowest one so that all the other degrees of freedom can easily equilibrate along the reaction coordinate. We used a regularised sparse autoencoder, an energy-based model, to discover a crucial set of reaction coordinates. Along with discovering reaction coordinates, our model also predicts the evolution of a molecular dynamics(MD) trajectory. We showcased that including sparsity enforcing regularisation helps in choosing a small but important set of reaction coordinates. We used two model systems to demonstrate our approach: alanine dipeptide system and proflavine and DNA system, which exhibited intercalation of proflavine into DNA minor groove in an aqueous environment. We model MD trajectory as a multivariate time series, and our latent variable model performs the task of multi-step time series prediction. This idea is inspired by the popular sparse coding approach - to represent each input sample as a linear combination of few elements taken from a set of representative patterns.
ESPRIT: Explaining Solutions to Physical Reasoning Tasks
May 14, 2020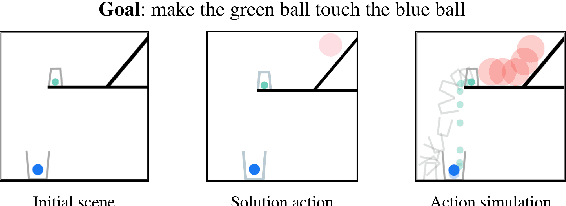

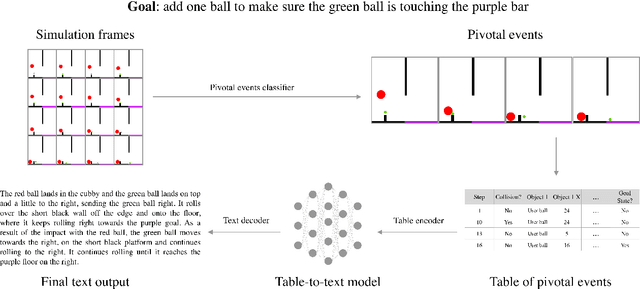
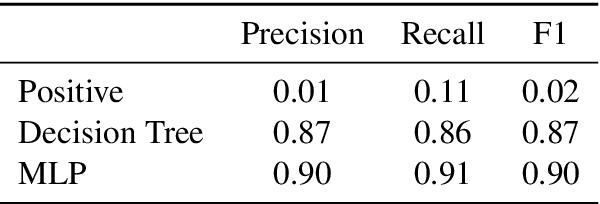
Neural networks lack the ability to reason about qualitative physics and so cannot generalize to scenarios and tasks unseen during training. We propose ESPRIT, a framework for commonsense reasoning about qualitative physics in natural language that generates interpretable descriptions of physical events. We use a two-step approach of first identifying the pivotal physical events in an environment and then generating natural language descriptions of those events using a data-to-text approach. Our framework learns to generate explanations of how the physical simulation will causally evolve so that an agent or a human can easily reason about a solution using those interpretable descriptions. Human evaluations indicate that ESPRIT produces crucial fine-grained details and has high coverage of physical concepts compared to even human annotations. Dataset, code and documentation are available at https://github.com/salesforce/esprit.