 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFLsim: A Modular and Library-Agnostic Simulation Framework for Federated Learning
Jul 15, 2025


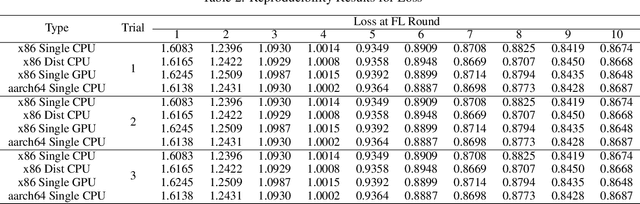
Federated Learning (FL) has undergone significant development since its inception in 2016, advancing from basic algorithms to complex methodologies tailored to address diverse challenges and use cases. However, research and benchmarking of novel FL techniques against a plethora of established state-of-the-art solutions remain challenging. To streamline this process, we introduce FLsim, a comprehensive FL simulation framework designed to meet the diverse requirements of FL workflows in the literature. FLsim is characterized by its modularity, scalability, resource efficiency, and controlled reproducibility of experimental outcomes. Its easy to use interface allows users to specify customized FL requirements through job configuration, which supports: (a) customized data distributions, ranging from non-independent and identically distributed (non-iid) data to independent and identically distributed (iid) data, (b) selection of local learning algorithms according to user preferences, with complete agnosticism to ML libraries, (c) choice of network topology illustrating communication patterns among nodes, (d) definition of model aggregation and consensus algorithms, and (e) pluggable blockchain support for enhanced robustness. Through a series of experimental evaluations, we demonstrate the effectiveness and versatility of FLsim in simulating a diverse range of state-of-the-art FL experiments. We envisage that FLsim would mark a significant advancement in FL simulation frameworks, offering unprecedented flexibility and functionality for researchers and practitioners alike.
CardiGraphormer: Unveiling the Power of Self-Supervised Learning in Revolutionizing Drug Discovery
Jul 04, 2023In the expansive realm of drug discovery, with approximately 15,000 known drugs and only around 4,200 approved, the combinatorial nature of the chemical space presents a formidable challenge. While Artificial Intelligence (AI) has emerged as a powerful ally, traditional AI frameworks face significant hurdles. This manuscript introduces CardiGraphormer, a groundbreaking approach that synergizes self-supervised learning (SSL), Graph Neural Networks (GNNs), and Cardinality Preserving Attention to revolutionize drug discovery. CardiGraphormer, a novel combination of Graphormer and Cardinality Preserving Attention, leverages SSL to learn potent molecular representations and employs GNNs to extract molecular fingerprints, enhancing predictive performance and interpretability while reducing computation time. It excels in handling complex data like molecular structures and performs tasks associated with nodes, pairs of nodes, subgraphs, or entire graph structures. CardiGraphormer's potential applications in drug discovery and drug interactions are vast, from identifying new drug targets to predicting drug-to-drug interactions and enabling novel drug discovery. This innovative approach provides an AI-enhanced methodology in drug development, utilizing SSL combined with GNNs to overcome existing limitations and pave the way for a richer exploration of the vast combinatorial chemical space in drug discovery.
Prediction of good reaction coordinates and future evolution of MD trajectories using Regularized Sparse Autoencoders: A novel deep learning approach
Aug 22, 2022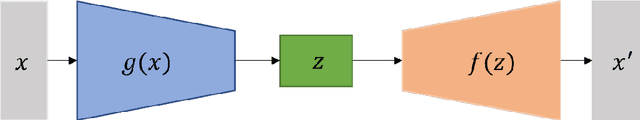

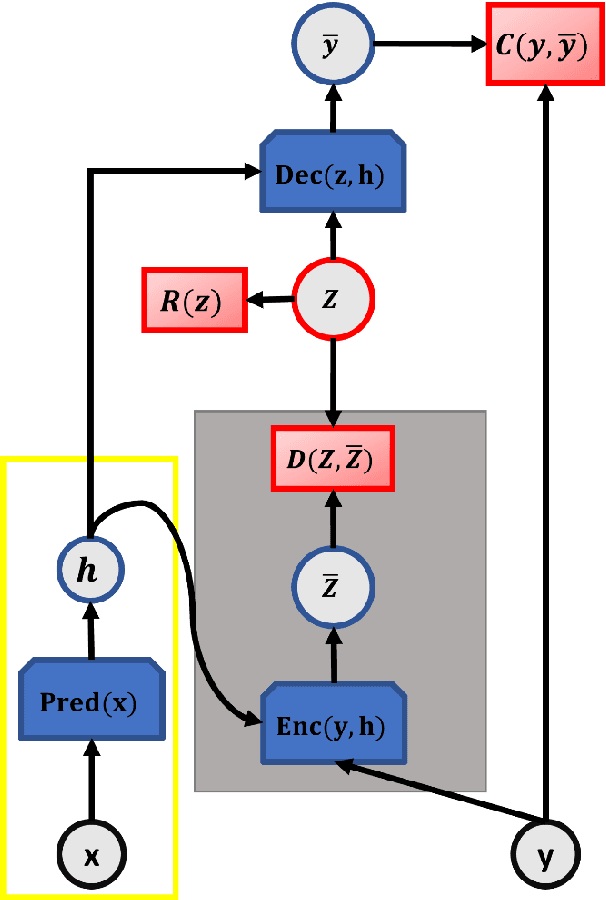
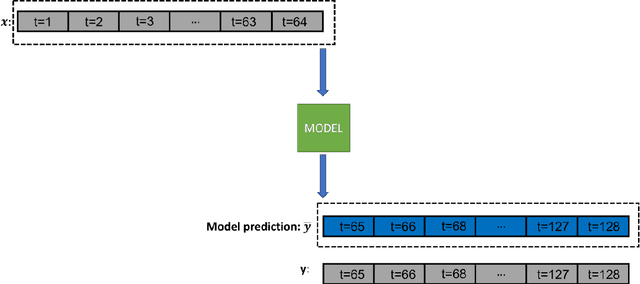
Identifying reaction coordinates(RCs) is an active area of research, given the crucial role RCs play in determining the progress of a chemical reaction. The choice of the reaction coordinate is often based on heuristic knowledge. However, an essential criterion for the choice is that the coordinate should capture both the reactant and product states unequivocally. Also, the coordinate should be the slowest one so that all the other degrees of freedom can easily equilibrate along the reaction coordinate. Also, the coordinate should be the slowest one so that all the other degrees of freedom can easily equilibrate along the reaction coordinate. We used a regularised sparse autoencoder, an energy-based model, to discover a crucial set of reaction coordinates. Along with discovering reaction coordinates, our model also predicts the evolution of a molecular dynamics(MD) trajectory. We showcased that including sparsity enforcing regularisation helps in choosing a small but important set of reaction coordinates. We used two model systems to demonstrate our approach: alanine dipeptide system and proflavine and DNA system, which exhibited intercalation of proflavine into DNA minor groove in an aqueous environment. We model MD trajectory as a multivariate time series, and our latent variable model performs the task of multi-step time series prediction. This idea is inspired by the popular sparse coding approach - to represent each input sample as a linear combination of few elements taken from a set of representative patterns.