 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGit-Theta: A Git Extension for Collaborative Development of Machine Learning Models
Jun 07, 2023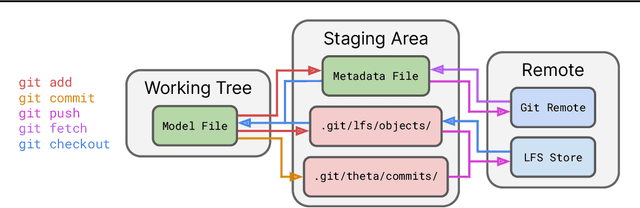
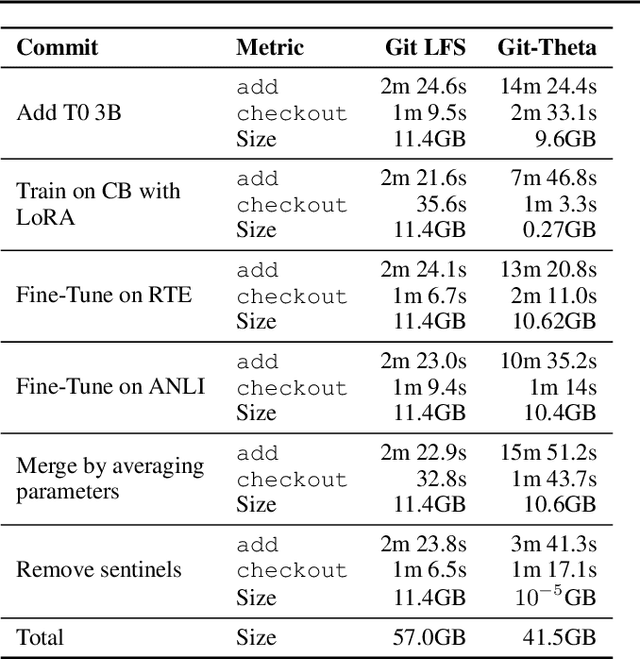
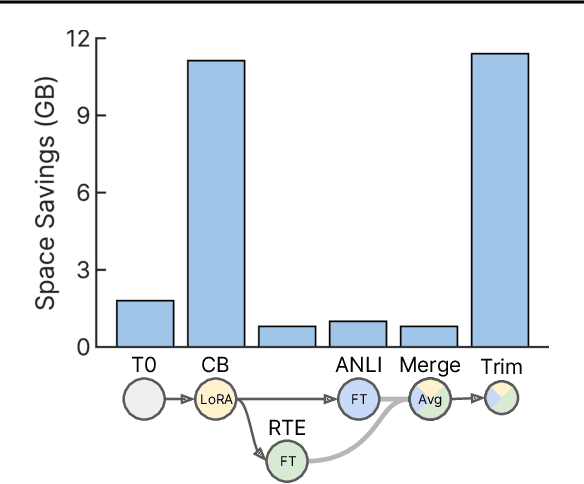
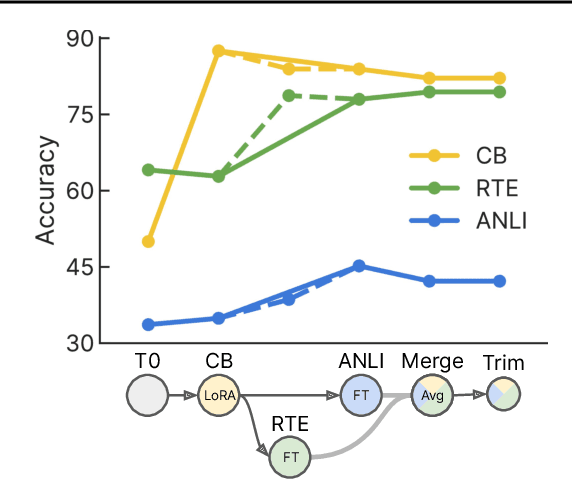
Currently, most machine learning models are trained by centralized teams and are rarely updated. In contrast, open-source software development involves the iterative development of a shared artifact through distributed collaboration using a version control system. In the interest of enabling collaborative and continual improvement of machine learning models, we introduce Git-Theta, a version control system for machine learning models. Git-Theta is an extension to Git, the most widely used version control software, that allows fine-grained tracking of changes to model parameters alongside code and other artifacts. Unlike existing version control systems that treat a model checkpoint as a blob of data, Git-Theta leverages the structure of checkpoints to support communication-efficient updates, automatic model merges, and meaningful reporting about the difference between two versions of a model. In addition, Git-Theta includes a plug-in system that enables users to easily add support for new functionality. In this paper, we introduce Git-Theta's design and features and include an example use-case of Git-Theta where a pre-trained model is continually adapted and modified. We publicly release Git-Theta in hopes of kickstarting a new era of collaborative model development.
Interpretable Single-Cell Set Classification with Kernel Mean Embeddings
Feb 10, 2022



Modern single-cell flow and mass cytometry technologies measure the expression of several proteins of the individual cells within a blood or tissue sample. Each profiled biological sample is thus represented by a set of hundreds of thousands of multidimensional cell feature vectors, which incurs a high computational cost to predict each biological sample's associated phenotype with machine learning models. Such a large set cardinality also limits the interpretability of machine learning models due to the difficulty in tracking how each individual cell influences the ultimate prediction. Using Kernel Mean Embedding to encode the cellular landscape of each profiled biological sample, we can train a simple linear classifier and achieve state-of-the-art classification accuracy on 3 flow and mass cytometry datasets. Our model contains few parameters but still performs similarly to deep learning models with millions of parameters. In contrast with deep learning approaches, the linearity and sub-selection step of our model make it easy to interpret classification results. Clustering analysis further shows that our method admits rich biological interpretability for linking cellular heterogeneity to clinical phenotype.