 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConditional Similarity Triplets Enable Covariate-Informed Representations of Single-Cell Data
Jun 12, 2024



Single-cell technologies enable comprehensive profiling of diverse immune cell-types through the measurement of multiple genes or proteins per cell. In order to translate data from immune profiling assays into powerful diagnostics, machine learning approaches are used to compute per-sample immunological summaries, or featurizations that can be used as inputs to models for outcomes of interest. Current supervised learning approaches for computing per-sample representations are optimized based only on the outcome variable to be predicted and do not take into account clinically-relevant covariates that are likely to also be measured. Here we expand the optimization problem to also take into account such additional patient covariates to directly inform the learned per-sample representations. To do this, we introduce CytoCoSet, a set-based encoding method, which formulates a loss function with an additional triplet term penalizing samples with similar covariates from having disparate embedding results in per-sample representations. Overall, incorporating clinical covariates leads to improved prediction of clinical phenotypes.
Interpretable Single-Cell Set Classification with Kernel Mean Embeddings
Feb 10, 2022



Modern single-cell flow and mass cytometry technologies measure the expression of several proteins of the individual cells within a blood or tissue sample. Each profiled biological sample is thus represented by a set of hundreds of thousands of multidimensional cell feature vectors, which incurs a high computational cost to predict each biological sample's associated phenotype with machine learning models. Such a large set cardinality also limits the interpretability of machine learning models due to the difficulty in tracking how each individual cell influences the ultimate prediction. Using Kernel Mean Embedding to encode the cellular landscape of each profiled biological sample, we can train a simple linear classifier and achieve state-of-the-art classification accuracy on 3 flow and mass cytometry datasets. Our model contains few parameters but still performs similarly to deep learning models with millions of parameters. In contrast with deep learning approaches, the linearity and sub-selection step of our model make it easy to interpret classification results. Clustering analysis further shows that our method admits rich biological interpretability for linking cellular heterogeneity to clinical phenotype.
Exchangeable Neural ODE for Set Modeling
Aug 06, 2020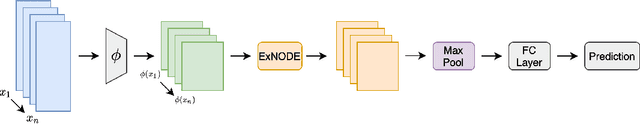


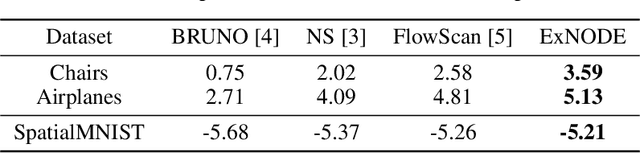
Reasoning over an instance composed of a set of vectors, like a point cloud, requires that one accounts for intra-set dependent features among elements. However, since such instances are unordered, the elements' features should remain unchanged when the input's order is permuted. This property, permutation equivariance, is a challenging constraint for most neural architectures. While recent work has proposed global pooling and attention-based solutions, these may be limited in the way that intradependencies are captured in practice. In this work we propose a more general formulation to achieve permutation equivariance through ordinary differential equations (ODE). Our proposed module, Exchangeable Neural ODE (ExNODE), can be seamlessly applied for both discriminative and generative tasks. We also extend set modeling in the temporal dimension and propose a VAE based model for temporal set modeling. Extensive experiments demonstrate the efficacy of our method over strong baselines.