 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDistribution-based Sketching of Single-Cell Samples
Jun 30, 2022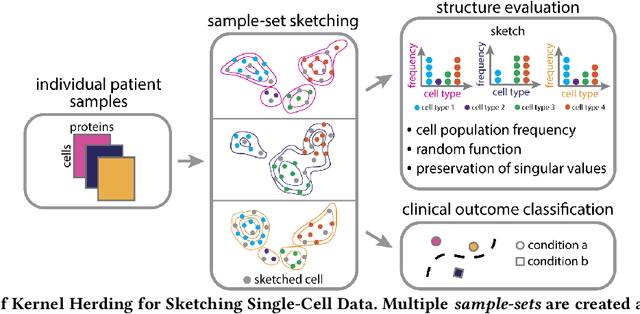
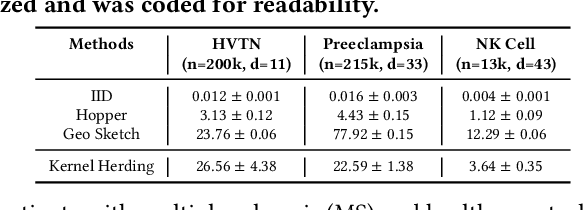
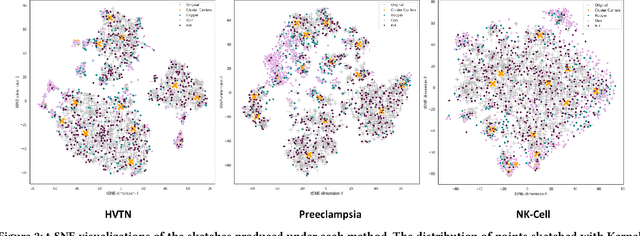
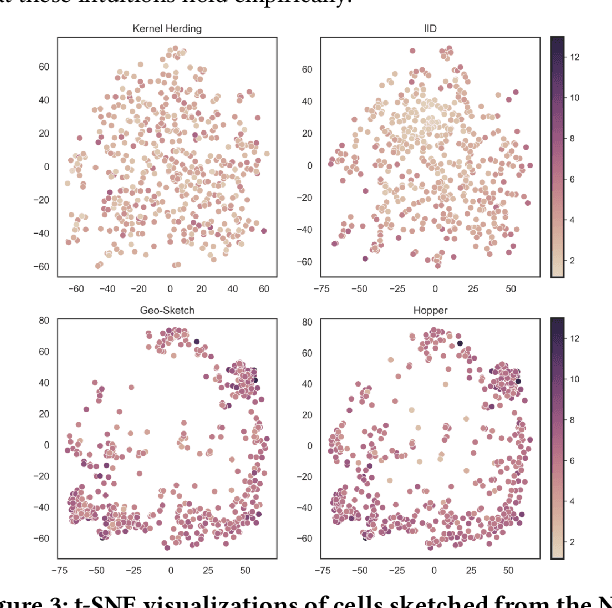
Modern high-throughput single-cell immune profiling technologies, such as flow and mass cytometry and single-cell RNA sequencing can readily measure the expression of a large number of protein or gene features across the millions of cells in a multi-patient cohort. While bioinformatics approaches can be used to link immune cell heterogeneity to external variables of interest, such as, clinical outcome or experimental label, they often struggle to accommodate such a large number of profiled cells. To ease this computational burden, a limited number of cells are typically \emph{sketched} or subsampled from each patient. However, existing sketching approaches fail to adequately subsample rare cells from rare cell-populations, or fail to preserve the true frequencies of particular immune cell-types. Here, we propose a novel sketching approach based on Kernel Herding that selects a limited subsample of all cells while preserving the underlying frequencies of immune cell-types. We tested our approach on three flow and mass cytometry datasets and on one single-cell RNA sequencing dataset and demonstrate that the sketched cells (1) more accurately represent the overall cellular landscape and (2) facilitate increased performance in downstream analysis tasks, such as classifying patients according to their clinical outcome. An implementation of sketching with Kernel Herding is publicly available at \url{https://github.com/vishalathreya/Set-Summarization}.
Interpretable Single-Cell Set Classification with Kernel Mean Embeddings
Feb 10, 2022



Modern single-cell flow and mass cytometry technologies measure the expression of several proteins of the individual cells within a blood or tissue sample. Each profiled biological sample is thus represented by a set of hundreds of thousands of multidimensional cell feature vectors, which incurs a high computational cost to predict each biological sample's associated phenotype with machine learning models. Such a large set cardinality also limits the interpretability of machine learning models due to the difficulty in tracking how each individual cell influences the ultimate prediction. Using Kernel Mean Embedding to encode the cellular landscape of each profiled biological sample, we can train a simple linear classifier and achieve state-of-the-art classification accuracy on 3 flow and mass cytometry datasets. Our model contains few parameters but still performs similarly to deep learning models with millions of parameters. In contrast with deep learning approaches, the linearity and sub-selection step of our model make it easy to interpret classification results. Clustering analysis further shows that our method admits rich biological interpretability for linking cellular heterogeneity to clinical phenotype.