 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Comprehensive Benchmark for RNA 3D Structure-Function Modeling
Mar 27, 2025


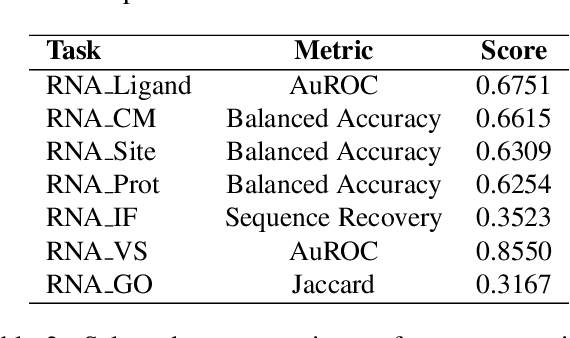
The RNA structure-function relationship has recently garnered significant attention within the deep learning community, promising to grow in importance as nucleic acid structure models advance. However, the absence of standardized and accessible benchmarks for deep learning on RNA 3D structures has impeded the development of models for RNA functional characteristics. In this work, we introduce a set of seven benchmarking datasets for RNA structure-function prediction, designed to address this gap. Our library builds on the established Python library rnaglib, and offers easy data distribution and encoding, splitters and evaluation methods, providing a convenient all-in-one framework for comparing models. Datasets are implemented in a fully modular and reproducible manner, facilitating for community contributions and customization. Finally, we provide initial baseline results for all tasks using a graph neural network. Source code: https://github.com/cgoliver/rnaglib Documentation: https://rnaglib.org
3D-based RNA function prediction tools in rnaglib
Feb 14, 2024Understanding the connection between complex structural features of RNA and biological function is a fundamental challenge in evolutionary studies and in RNA design. However, building datasets of RNA 3D structures and making appropriate modeling choices remains time-consuming and lacks standardization. In this chapter, we describe the use of rnaglib, to train supervised and unsupervised machine learning-based function prediction models on datasets of RNA 3D structures.
AtomSurf : Surface Representation for Learning on Protein Structures
Sep 28, 2023



Recent advancements in Cryo-EM and protein structure prediction algorithms have made large-scale protein structures accessible, paving the way for machine learning-based functional annotations.The field of geometric deep learning focuses on creating methods working on geometric data. An essential aspect of learning from protein structures is representing these structures as a geometric object (be it a grid, graph, or surface) and applying a learning method tailored to this representation. The performance of a given approach will then depend on both the representation and its corresponding learning method. In this paper, we investigate representing proteins as $\textit{3D mesh surfaces}$ and incorporate them into an established representation benchmark. Our first finding is that despite promising preliminary results, the surface representation alone does not seem competitive with 3D grids. Building on this, we introduce a synergistic approach, combining surface representations with graph-based methods, resulting in a general framework that incorporates both representations in learning. We show that using this combination, we are able to obtain state-of-the-art results across $\textit{all tested tasks}$. Our code and data can be found online: https://github.com/Vincentx15/atom2D .
Approximate Network Motif Mining Via Graph Learning
Jun 07, 2022

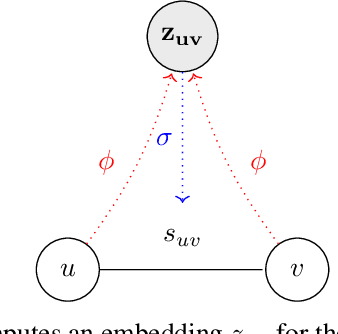

Frequent and structurally related subgraphs, also known as network motifs, are valuable features of many graph datasets. However, the high computational complexity of identifying motif sets in arbitrary datasets (motif mining) has limited their use in many real-world datasets. By automatically leveraging statistical properties of datasets, machine learning approaches have shown promise in several tasks with combinatorial complexity and are therefore a promising candidate for network motif mining. In this work we seek to facilitate the development of machine learning approaches aimed at motif mining. We propose a formulation of the motif mining problem as a node labelling task. In addition, we build benchmark datasets and evaluation metrics which test the ability of models to capture different aspects of motif discovery such as motif number, size, topology, and scarcity. Next, we propose MotiFiesta, a first attempt at solving this problem in a fully differentiable manner with promising results on challenging baselines. Finally, we demonstrate through MotiFiesta that this learning setting can be applied simultaneously to general-purpose data mining and interpretable feature extraction for graph classification tasks.
Edge-similarity-aware Graph Neural Networks
Sep 20, 2021
Graph are a ubiquitous data representation, as they represent a flexible and compact representation. For instance, the 3D structure of RNA can be efficiently represented as $\textit{2.5D graphs}$, graphs whose nodes are nucleotides and edges represent chemical interactions. In this setting, we have biological evidence of the similarity between the edge types, as some chemical interactions are more similar than others. Machine learning on graphs have recently experienced a breakthrough with the introduction of Graph Neural Networks. This algorithm can be framed as a message passing algorithm between graph nodes over graph edges. These messages can depend on the edge type they are transmitted through, but no method currently constrains how a message is altered when the edge type changes. Motivated by the RNA use case, in this project we introduce a graph neural network layer which can leverage prior information about similarities between edges. We show that despite the theoretical appeal of including this similarity prior, the empirical performance is not enhanced on the tasks and datasets we include here.
VeRNAl: A Tool for Mining Fuzzy Network Motifs in RNA
Sep 01, 2020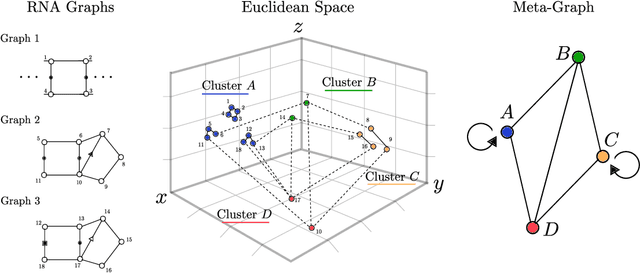


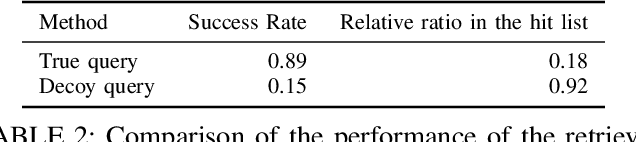
Motivation: RNAs are ubiquitous molecules involved in many regulatory and catalytic processes. Their ability to form complex structures is often key to support these functions. Remarkably, RNA 3D structures are articulated around smaller 3D sub-units referred as RNA 3D motifs that can be found in unrelated molecules. The classification of these 3D motifs is thus essential to characterize RNA structures, but current methods can only retrieve motifs with identical base interaction patterns. Results: Here, we relax this constraint by posing the motif finding problem as a graph representation learning and clustering task. This framing takes advantage of the continuous nature of graph representations to model the flexibility of RNA motifs while retaining the convenient encoding of RNAs as graphs. We propose a set of node similarity functions, clustering methods, and motif construction algorithms to recover flexible RNA motifs. We show that our methods are able to retrieve and expand known classes of motifs, but also to identify new motifs. Our tool, VeRNAl can be easily customized by users to desired levels of motif flexibility, abundance and size. Availability and Implementation: The source code, data, and a webserver are available at vernal.cs.mcgill.ca
Leveraging binding-site structure for drug discovery with point-cloud methods
May 28, 2019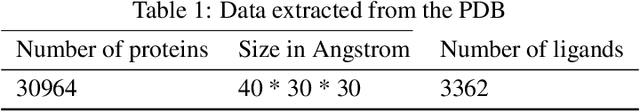


Computational drug discovery strategies can be broadly placed in two categories: ligand-based methods which identify novel molecules by similarity with known ligands, and structure-based methods which predict molecules with high-affinity to a given 3D structure (e.g. a protein). However, ligand-based methods do not leverage information about the binding site, and structure-based approaches rely on the knowledge of a finite set of ligands binding the target. In this work, we introduce TarLig, a novel approach that aims to bridge the gap between ligand and structure-based approaches. We use the 3D structure of the binding site as input to a model which predicts the ligand preferences of the binding site. The resulting predictions could then offer promising seeds and constraints in the chemical space search, based on the binding site structure. TarLig outperforms standard models by introducing a data-alignment and augmentation technique. The recent popularity of Volumetric 3DCNN pipelines in structural bioinformatics suggests that this extra step could help a wide range of methods to improve their results with minimal modifications.