 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge14 Examples of How LLMs Can Transform Materials Science and Chemistry: A Reflection on a Large Language Model Hackathon
Jun 13, 2023



Chemistry and materials science are complex. Recently, there have been great successes in addressing this complexity using data-driven or computational techniques. Yet, the necessity of input structured in very specific forms and the fact that there is an ever-growing number of tools creates usability and accessibility challenges. Coupled with the reality that much data in these disciplines is unstructured, the effectiveness of these tools is limited. Motivated by recent works that indicated that large language models (LLMs) might help address some of these issues, we organized a hackathon event on the applications of LLMs in chemistry, materials science, and beyond. This article chronicles the projects built as part of this hackathon. Participants employed LLMs for various applications, including predicting properties of molecules and materials, designing novel interfaces for tools, extracting knowledge from unstructured data, and developing new educational applications. The diverse topics and the fact that working prototypes could be generated in less than two days highlight that LLMs will profoundly impact the future of our fields. The rich collection of ideas and projects also indicates that the applications of LLMs are not limited to materials science and chemistry but offer potential benefits to a wide range of scientific disciplines.
Leveraging binding-site structure for drug discovery with point-cloud methods
May 28, 2019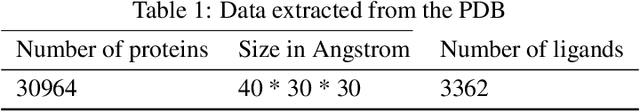


Computational drug discovery strategies can be broadly placed in two categories: ligand-based methods which identify novel molecules by similarity with known ligands, and structure-based methods which predict molecules with high-affinity to a given 3D structure (e.g. a protein). However, ligand-based methods do not leverage information about the binding site, and structure-based approaches rely on the knowledge of a finite set of ligands binding the target. In this work, we introduce TarLig, a novel approach that aims to bridge the gap between ligand and structure-based approaches. We use the 3D structure of the binding site as input to a model which predicts the ligand preferences of the binding site. The resulting predictions could then offer promising seeds and constraints in the chemical space search, based on the binding site structure. TarLig outperforms standard models by introducing a data-alignment and augmentation technique. The recent popularity of Volumetric 3DCNN pipelines in structural bioinformatics suggests that this extra step could help a wide range of methods to improve their results with minimal modifications.