 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVeRNAl: A Tool for Mining Fuzzy Network Motifs in RNA
Sep 01, 2020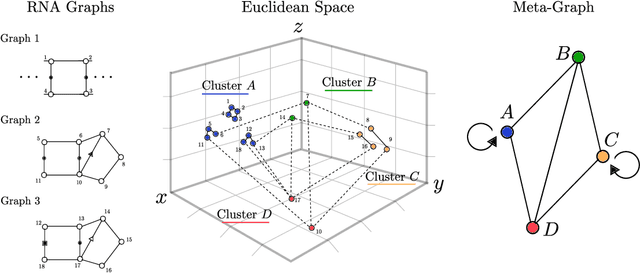


Motivation: RNAs are ubiquitous molecules involved in many regulatory and catalytic processes. Their ability to form complex structures is often key to support these functions. Remarkably, RNA 3D structures are articulated around smaller 3D sub-units referred as RNA 3D motifs that can be found in unrelated molecules. The classification of these 3D motifs is thus essential to characterize RNA structures, but current methods can only retrieve motifs with identical base interaction patterns. Results: Here, we relax this constraint by posing the motif finding problem as a graph representation learning and clustering task. This framing takes advantage of the continuous nature of graph representations to model the flexibility of RNA motifs while retaining the convenient encoding of RNAs as graphs. We propose a set of node similarity functions, clustering methods, and motif construction algorithms to recover flexible RNA motifs. We show that our methods are able to retrieve and expand known classes of motifs, but also to identify new motifs. Our tool, VeRNAl can be easily customized by users to desired levels of motif flexibility, abundance and size. Availability and Implementation: The source code, data, and a webserver are available at vernal.cs.mcgill.ca
Leveraging binding-site structure for drug discovery with point-cloud methods
May 28, 2019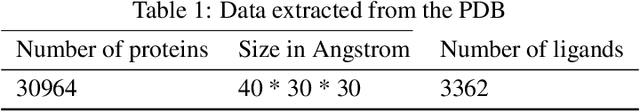


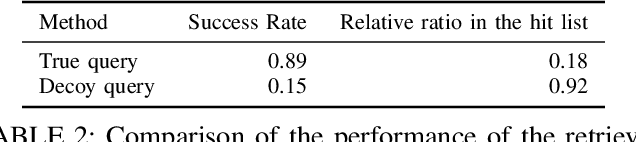
Computational drug discovery strategies can be broadly placed in two categories: ligand-based methods which identify novel molecules by similarity with known ligands, and structure-based methods which predict molecules with high-affinity to a given 3D structure (e.g. a protein). However, ligand-based methods do not leverage information about the binding site, and structure-based approaches rely on the knowledge of a finite set of ligands binding the target. In this work, we introduce TarLig, a novel approach that aims to bridge the gap between ligand and structure-based approaches. We use the 3D structure of the binding site as input to a model which predicts the ligand preferences of the binding site. The resulting predictions could then offer promising seeds and constraints in the chemical space search, based on the binding site structure. TarLig outperforms standard models by introducing a data-alignment and augmentation technique. The recent popularity of Volumetric 3DCNN pipelines in structural bioinformatics suggests that this extra step could help a wide range of methods to improve their results with minimal modifications.