 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBioBlobs: Differentiable Graph Partitioning for Protein Representation Learning
Oct 02, 2025Protein function is driven by coherent substructures which vary in size and topology, yet current protein representation learning models (PRL) distort these signals by relying on rigid substructures such as k-hop and fixed radius neighbourhoods. We introduce BioBlobs, a plug-and-play, fully differentiable module that represents proteins by dynamically partitioning structures into flexibly-sized, non-overlapping substructures ("blobs"). The resulting blobs are quantized into a shared and interpretable codebook, yielding a discrete vocabulary of function-relevant protein substructures used to compute protein embeddings. We show that BioBlobs representations improve the performance of widely used protein encoders such as GVP-GNN across various PRL tasks. Our approach highlights the value of architectures that directly capture function-relevant protein substructures, enabling both improved predictive performance and mechanistic insight into protein function.
A Comprehensive Benchmark for RNA 3D Structure-Function Modeling
Mar 27, 2025


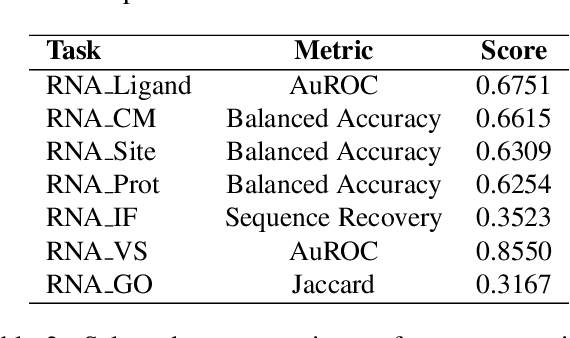
The RNA structure-function relationship has recently garnered significant attention within the deep learning community, promising to grow in importance as nucleic acid structure models advance. However, the absence of standardized and accessible benchmarks for deep learning on RNA 3D structures has impeded the development of models for RNA functional characteristics. In this work, we introduce a set of seven benchmarking datasets for RNA structure-function prediction, designed to address this gap. Our library builds on the established Python library rnaglib, and offers easy data distribution and encoding, splitters and evaluation methods, providing a convenient all-in-one framework for comparing models. Datasets are implemented in a fully modular and reproducible manner, facilitating for community contributions and customization. Finally, we provide initial baseline results for all tasks using a graph neural network. Source code: https://github.com/cgoliver/rnaglib Documentation: https://rnaglib.org
3D-based RNA function prediction tools in rnaglib
Feb 14, 2024Understanding the connection between complex structural features of RNA and biological function is a fundamental challenge in evolutionary studies and in RNA design. However, building datasets of RNA 3D structures and making appropriate modeling choices remains time-consuming and lacks standardization. In this chapter, we describe the use of rnaglib, to train supervised and unsupervised machine learning-based function prediction models on datasets of RNA 3D structures.
Endowing Protein Language Models with Structural Knowledge
Jan 26, 2024Understanding the relationships between protein sequence, structure and function is a long-standing biological challenge with manifold implications from drug design to our understanding of evolution. Recently, protein language models have emerged as the preferred method for this challenge, thanks to their ability to harness large sequence databases. Yet, their reliance on expansive sequence data and parameter sets limits their flexibility and practicality in real-world scenarios. Concurrently, the recent surge in computationally predicted protein structures unlocks new opportunities in protein representation learning. While promising, the computational burden carried by such complex data still hinders widely-adopted practical applications. To address these limitations, we introduce a novel framework that enhances protein language models by integrating protein structural data. Drawing from recent advances in graph transformers, our approach refines the self-attention mechanisms of pretrained language transformers by integrating structural information with structure extractor modules. This refined model, termed Protein Structure Transformer (PST), is further pretrained on a small protein structure database, using the same masked language modeling objective as traditional protein language models. Empirical evaluations of PST demonstrate its superior parameter efficiency relative to protein language models, despite being pretrained on a dataset comprising only 542K structures. Notably, PST consistently outperforms the state-of-the-art foundation model for protein sequences, ESM-2, setting a new benchmark in protein function prediction. Our findings underscore the potential of integrating structural information into protein language models, paving the way for more effective and efficient protein modeling Code and pretrained models are available at https://github.com/BorgwardtLab/PST.
Unsupervised Manifold Alignment with Joint Multidimensional Scaling
Jul 06, 2022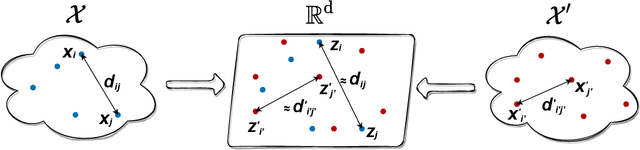
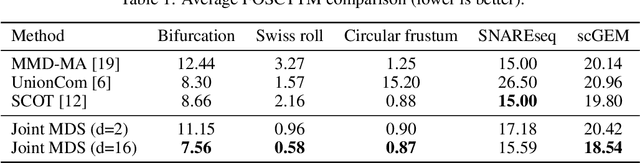
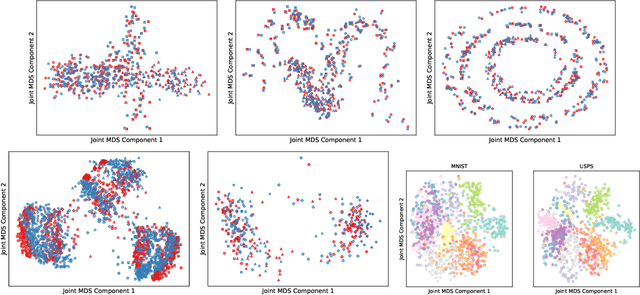

We introduce Joint Multidimensional Scaling, a novel approach for unsupervised manifold alignment, which maps datasets from two different domains, without any known correspondences between data instances across the datasets, to a common low-dimensional Euclidean space. Our approach integrates Multidimensional Scaling (MDS) and Wasserstein Procrustes analysis into a joint optimization problem to simultaneously generate isometric embeddings of data and learn correspondences between instances from two different datasets, while only requiring intra-dataset pairwise dissimilarities as input. This unique characteristic makes our approach applicable to datasets without access to the input features, such as solving the inexact graph matching problem. We propose an alternating optimization scheme to solve the problem that can fully benefit from the optimization techniques for MDS and Wasserstein Procrustes. We demonstrate the effectiveness of our approach in several applications, including joint visualization of two datasets, unsupervised heterogeneous domain adaptation, graph matching, and protein structure alignment.
Approximate Network Motif Mining Via Graph Learning
Jun 07, 2022

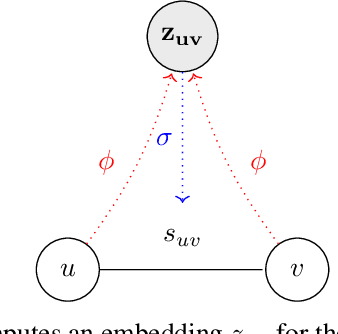

Frequent and structurally related subgraphs, also known as network motifs, are valuable features of many graph datasets. However, the high computational complexity of identifying motif sets in arbitrary datasets (motif mining) has limited their use in many real-world datasets. By automatically leveraging statistical properties of datasets, machine learning approaches have shown promise in several tasks with combinatorial complexity and are therefore a promising candidate for network motif mining. In this work we seek to facilitate the development of machine learning approaches aimed at motif mining. We propose a formulation of the motif mining problem as a node labelling task. In addition, we build benchmark datasets and evaluation metrics which test the ability of models to capture different aspects of motif discovery such as motif number, size, topology, and scarcity. Next, we propose MotiFiesta, a first attempt at solving this problem in a fully differentiable manner with promising results on challenging baselines. Finally, we demonstrate through MotiFiesta that this learning setting can be applied simultaneously to general-purpose data mining and interpretable feature extraction for graph classification tasks.
VeRNAl: A Tool for Mining Fuzzy Network Motifs in RNA
Sep 01, 2020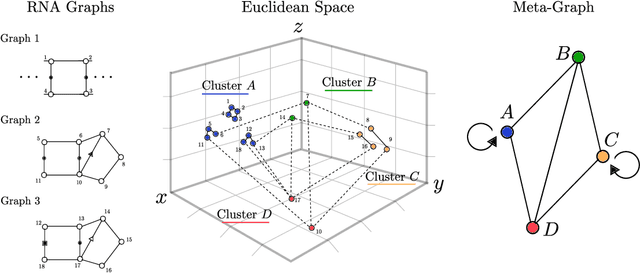


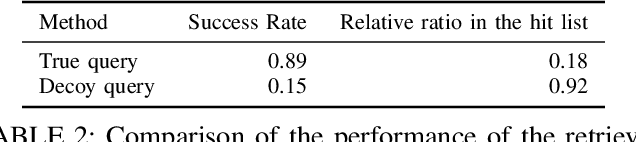
Motivation: RNAs are ubiquitous molecules involved in many regulatory and catalytic processes. Their ability to form complex structures is often key to support these functions. Remarkably, RNA 3D structures are articulated around smaller 3D sub-units referred as RNA 3D motifs that can be found in unrelated molecules. The classification of these 3D motifs is thus essential to characterize RNA structures, but current methods can only retrieve motifs with identical base interaction patterns. Results: Here, we relax this constraint by posing the motif finding problem as a graph representation learning and clustering task. This framing takes advantage of the continuous nature of graph representations to model the flexibility of RNA motifs while retaining the convenient encoding of RNAs as graphs. We propose a set of node similarity functions, clustering methods, and motif construction algorithms to recover flexible RNA motifs. We show that our methods are able to retrieve and expand known classes of motifs, but also to identify new motifs. Our tool, VeRNAl can be easily customized by users to desired levels of motif flexibility, abundance and size. Availability and Implementation: The source code, data, and a webserver are available at vernal.cs.mcgill.ca