 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDrug response prediction by inferring pathway-response associations with Kernelized Bayesian Matrix Factorization
Jun 11, 2016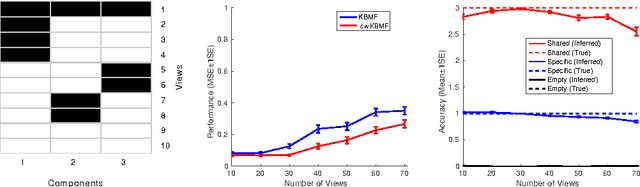
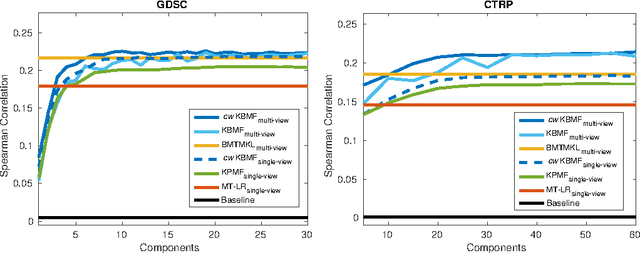
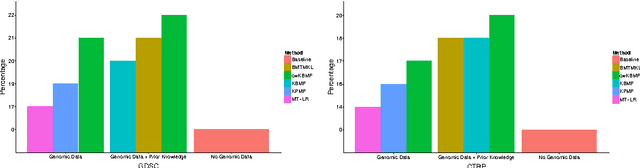
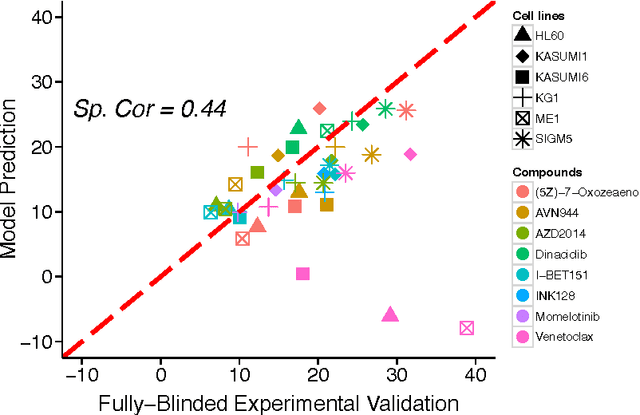
A key goal of computational personalized medicine is to systematically utilize genomic and other molecular features of samples to predict drug responses for a previously unseen sample. Such predictions are valuable for developing hypotheses for selecting therapies tailored for individual patients. This is especially valuable in oncology, where molecular and genetic heterogeneity of the cells has a major impact on the response. However, the prediction task is extremely challenging, raising the need for methods that can effectively model and predict drug responses. In this study, we propose a novel formulation of multi-task matrix factorization that allows selective data integration for predicting drug responses. To solve the modeling task, we extend the state-of-the-art kernelized Bayesian matrix factorization (KBMF) method with component-wise multiple kernel learning. In addition, our approach exploits the known pathway information in a novel and biologically meaningful fashion to learn the drug response associations. Our method quantitatively outperforms the state of the art on predicting drug responses in two publicly available cancer data sets as well as on a synthetic data set. In addition, we validated our model predictions with lab experiments using an in-house cancer cell line panel. We finally show the practical applicability of the proposed method by utilizing prior knowledge to infer pathway-drug response associations, opening up the opportunity for elucidating drug action mechanisms. We demonstrate that pathway-response associations can be learned by the proposed model for the well known EGFR and MEK inhibitors.
* Accepted in European Conference in Computational Biology, to be published in Bioinformatics 2016
A two-step learning approach for solving full and almost full cold start problems in dyadic prediction
May 17, 2014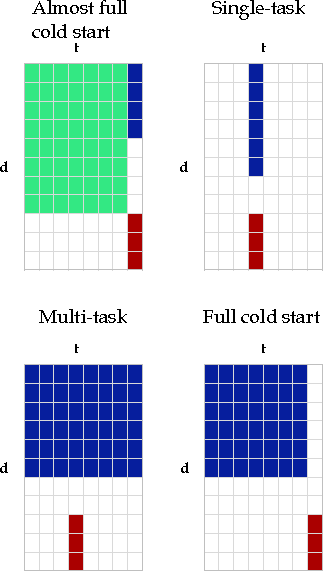
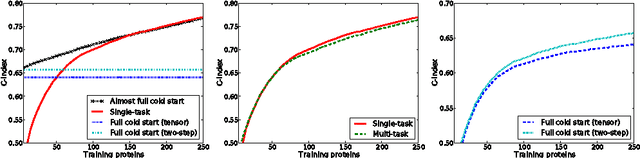
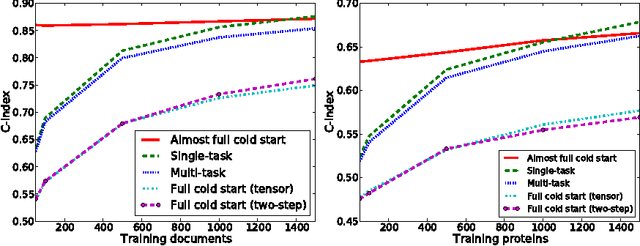
Dyadic prediction methods operate on pairs of objects (dyads), aiming to infer labels for out-of-sample dyads. We consider the full and almost full cold start problem in dyadic prediction, a setting that occurs when both objects in an out-of-sample dyad have not been observed during training, or if one of them has been observed, but very few times. A popular approach for addressing this problem is to train a model that makes predictions based on a pairwise feature representation of the dyads, or, in case of kernel methods, based on a tensor product pairwise kernel. As an alternative to such a kernel approach, we introduce a novel two-step learning algorithm that borrows ideas from the fields of pairwise learning and spectral filtering. We show theoretically that the two-step method is very closely related to the tensor product kernel approach, and experimentally that it yields a slightly better predictive performance. Moreover, unlike existing tensor product kernel methods, the two-step method allows closed-form solutions for training and parameter selection via cross-validation estimates both in the full and almost full cold start settings, making the approach much more efficient and straightforward to implement.
RPA: Probabilistic analysis of probe performance and robust summarization
Apr 06, 2013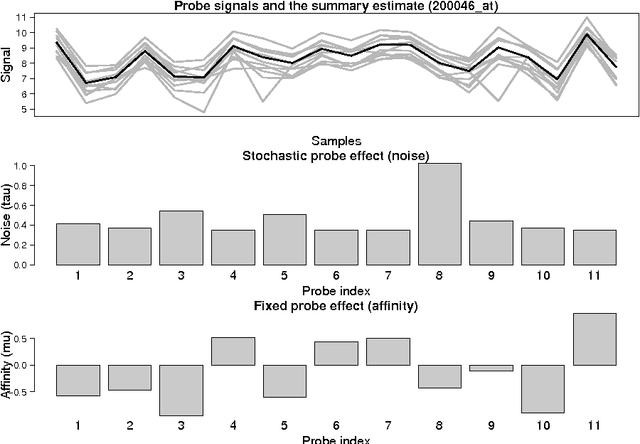
Probe-level models have led to improved performance in microarray studies but the various sources of probe-level contamination are still poorly understood. Data-driven analysis of probe performance can be used to quantify the uncertainty in individual probes and to highlight the relative contribution of different noise sources. Improved understanding of the probe-level effects can lead to improved preprocessing techniques and microarray design. We have implemented probabilistic tools for probe performance analysis and summarization on short oligonucleotide arrays. In contrast to standard preprocessing approaches, the methods provide quantitative estimates of probe-specific noise and affinity terms and tools to investigate these parameters. Tools to incorporate prior information of the probes in the analysis are provided as well. Comparisons to known probe-level error sources and spike-in data sets validate the approach. Implementation is freely available in R/BioConductor: http://www.bioconductor.org/packages/release/bioc/html/RPA.html