 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRPA: Probabilistic analysis of probe performance and robust summarization
Apr 06, 2013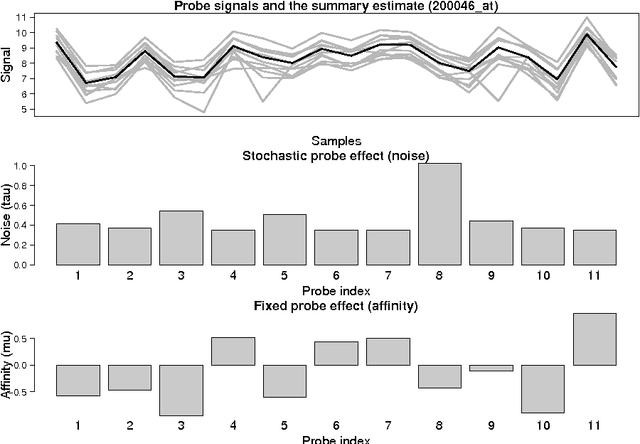
Probe-level models have led to improved performance in microarray studies but the various sources of probe-level contamination are still poorly understood. Data-driven analysis of probe performance can be used to quantify the uncertainty in individual probes and to highlight the relative contribution of different noise sources. Improved understanding of the probe-level effects can lead to improved preprocessing techniques and microarray design. We have implemented probabilistic tools for probe performance analysis and summarization on short oligonucleotide arrays. In contrast to standard preprocessing approaches, the methods provide quantitative estimates of probe-specific noise and affinity terms and tools to investigate these parameters. Tools to incorporate prior information of the probes in the analysis are provided as well. Comparisons to known probe-level error sources and spike-in data sets validate the approach. Implementation is freely available in R/BioConductor: http://www.bioconductor.org/packages/release/bioc/html/RPA.html
Fully scalable online-preprocessing algorithm for short oligonucleotide microarray atlases
Dec 27, 2012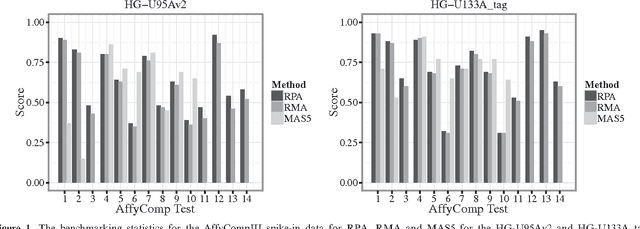
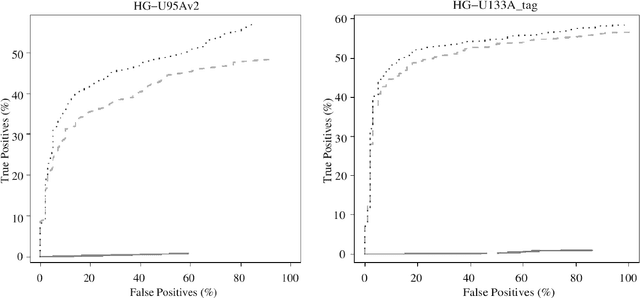
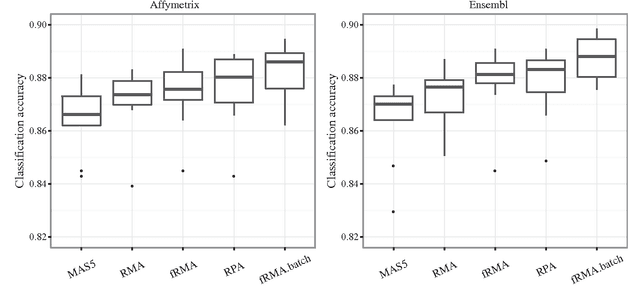
Accumulation of standardized data collections is opening up novel opportunities for holistic characterization of genome function. The limited scalability of current preprocessing techniques has, however, formed a bottleneck for full utilization of contemporary microarray collections. While short oligonucleotide arrays constitute a major source of genome-wide profiling data, scalable probe-level preprocessing algorithms have been available only for few measurement platforms based on pre-calculated model parameters from restricted reference training sets. To overcome these key limitations, we introduce a fully scalable online-learning algorithm that provides tools to process large microarray atlases including tens of thousands of arrays. Unlike the alternatives, the proposed algorithm scales up in linear time with respect to sample size and is readily applicable to all short oligonucleotide platforms. This is the only available preprocessing algorithm that can learn probe-level parameters based on sequential hyperparameter updates at small, consecutive batches of data, thus circumventing the extensive memory requirements of the standard approaches and opening up novel opportunities to take full advantage of contemporary microarray data collections. Moreover, using the most comprehensive data collections to estimate probe-level effects can assist in pinpointing individual probes affected by various biases and provide new tools to guide array design and quality control. The implementation is freely available in R/Bioconductor at http://www.bioconductor.org/packages/devel/bioc/html/RPA.html
* 20 pages, 3 figures, 1 supplementary PDF
Global modeling of transcriptional responses in interaction networks
Feb 02, 2012
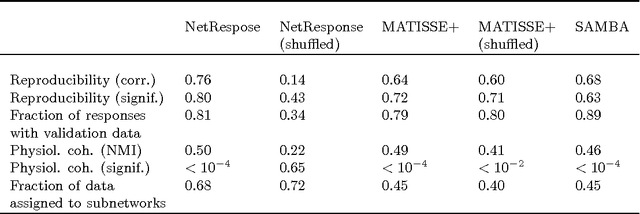


Motivation: Cell-biological processes are regulated through a complex network of interactions between genes and their products. The processes, their activating conditions, and the associated transcriptional responses are often unknown. Organism-wide modeling of network activation can reveal unique and shared mechanisms between physiological conditions, and potentially as yet unknown processes. We introduce a novel approach for organism-wide discovery and analysis of transcriptional responses in interaction networks. The method searches for local, connected regions in a network that exhibit coordinated transcriptional response in a subset of conditions. Known interactions between genes are used to limit the search space and to guide the analysis. Validation on a human pathway network reveals physiologically coherent responses, functional relatedness between physiological conditions, and coordinated, context-specific regulation of the genes. Availability: Implementation is freely available in R and Matlab at http://netpro.r-forge.r-project.org
* 19 pages, 13 figures
Probabilistic analysis of the human transcriptome with side information
Feb 27, 2011


Understanding functional organization of genetic information is a major challenge in modern biology. Following the initial publication of the human genome sequence in 2001, advances in high-throughput measurement technologies and efficient sharing of research material through community databases have opened up new views to the study of living organisms and the structure of life. In this thesis, novel computational strategies have been developed to investigate a key functional layer of genetic information, the human transcriptome, which regulates the function of living cells through protein synthesis. The key contributions of the thesis are general exploratory tools for high-throughput data analysis that have provided new insights to cell-biological networks, cancer mechanisms and other aspects of genome function. A central challenge in functional genomics is that high-dimensional genomic observations are associated with high levels of complex and largely unknown sources of variation. By combining statistical evidence across multiple measurement sources and the wealth of background information in genomic data repositories it has been possible to solve some the uncertainties associated with individual observations and to identify functional mechanisms that could not be detected based on individual measurement sources. Statistical learning and probabilistic models provide a natural framework for such modeling tasks. Open source implementations of the key methodological contributions have been released to facilitate further adoption of the developed methods by the research community.
* Doctoral thesis. 103 pages, 11 figures
Dependency detection with similarity constraints
Jan 31, 2011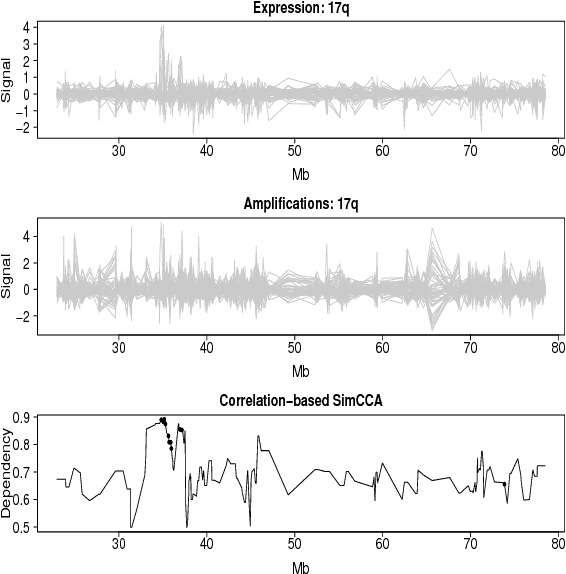
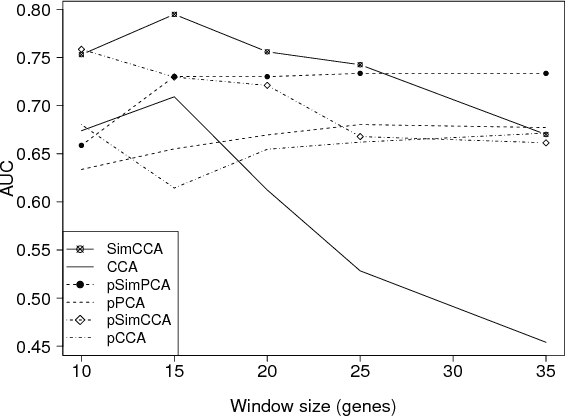
Unsupervised two-view learning, or detection of dependencies between two paired data sets, is typically done by some variant of canonical correlation analysis (CCA). CCA searches for a linear projection for each view, such that the correlations between the projections are maximized. The solution is invariant to any linear transformation of either or both of the views; for tasks with small sample size such flexibility implies overfitting, which is even worse for more flexible nonparametric or kernel-based dependency discovery methods. We develop variants which reduce the degrees of freedom by assuming constraints on similarity of the projections in the two views. A particular example is provided by a cancer gene discovery application where chromosomal distance affects the dependencies between gene copy number and activity levels. Similarity constraints are shown to improve detection performance of known cancer genes.
* 9 pages, 3 figures. Appeared in proceedings of the 2009 IEEE International Workshop on Machine Learning for Signal Processing XIX (MLSP'09). Implementation of the method available at http://bioconductor.org/packages/devel/bioc/html/pint.html