 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeToward computational cumulative biology by combining models of biological datasets
Apr 01, 2014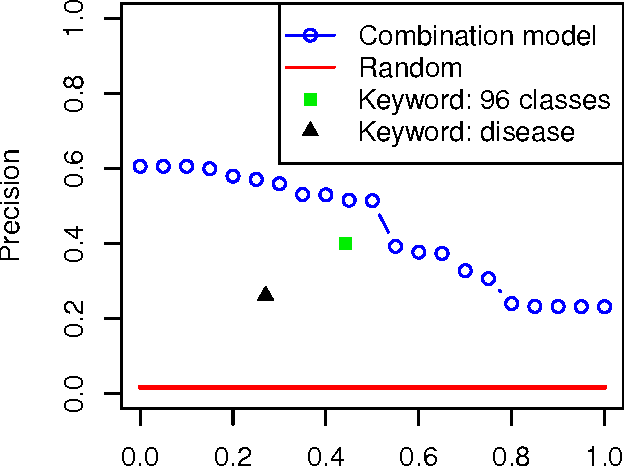
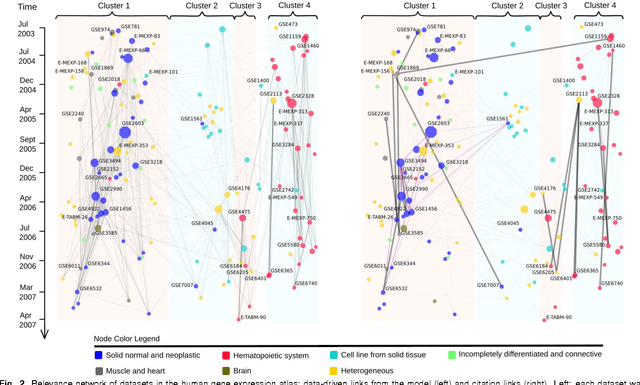
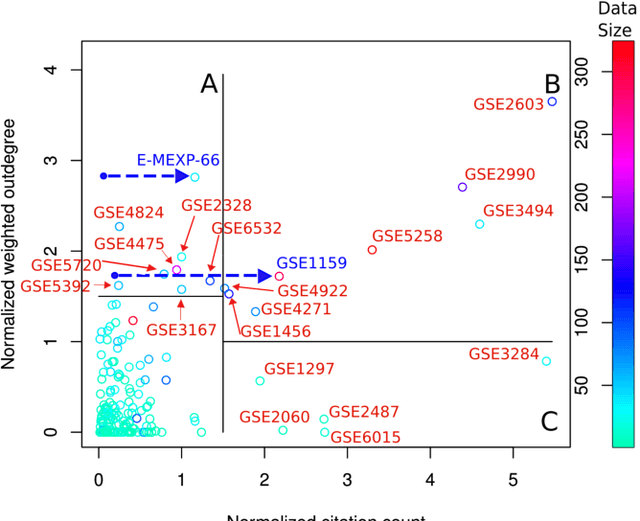
A main challenge of data-driven sciences is how to make maximal use of the progressively expanding databases of experimental datasets in order to keep research cumulative. We introduce the idea of a modeling-based dataset retrieval engine designed for relating a researcher's experimental dataset to earlier work in the field. The search is (i) data-driven to enable new findings, going beyond the state of the art of keyword searches in annotations, (ii) modeling-driven, to both include biological knowledge and insights learned from data, and (iii) scalable, as it is accomplished without building one unified grand model of all data. Assuming each dataset has been modeled beforehand, by the researchers or by database managers, we apply a rapidly computable and optimizable combination model to decompose a new dataset into contributions from earlier relevant models. By using the data-driven decomposition we identify a network of interrelated datasets from a large annotated human gene expression atlas. While tissue type and disease were major driving forces for determining relevant datasets, the found relationships were richer and the model-based search was more accurate than keyword search; it moreover recovered biologically meaningful relationships that are not straightforwardly visible from annotations, for instance, between cells in different developmental stages such as thymocytes and T-cells. Data-driven links and citations matched to a large extent; the data-driven links even uncovered corrections to the publication data, as two of the most linked datasets were not highly cited and turned out to have wrong publication entries in the database.
Fully scalable online-preprocessing algorithm for short oligonucleotide microarray atlases
Dec 27, 2012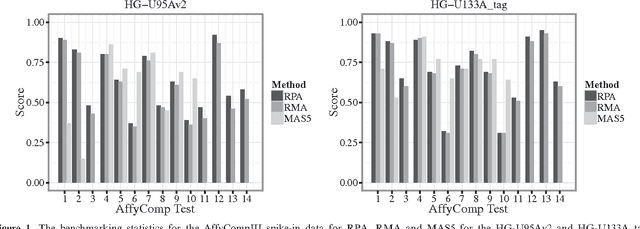
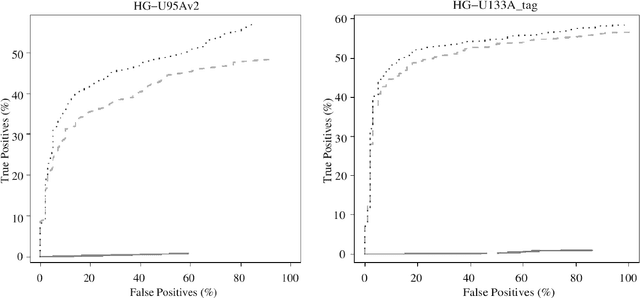
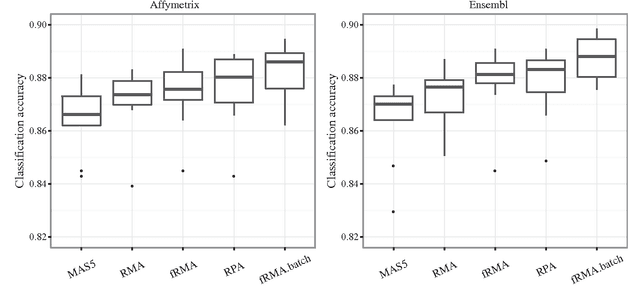
Accumulation of standardized data collections is opening up novel opportunities for holistic characterization of genome function. The limited scalability of current preprocessing techniques has, however, formed a bottleneck for full utilization of contemporary microarray collections. While short oligonucleotide arrays constitute a major source of genome-wide profiling data, scalable probe-level preprocessing algorithms have been available only for few measurement platforms based on pre-calculated model parameters from restricted reference training sets. To overcome these key limitations, we introduce a fully scalable online-learning algorithm that provides tools to process large microarray atlases including tens of thousands of arrays. Unlike the alternatives, the proposed algorithm scales up in linear time with respect to sample size and is readily applicable to all short oligonucleotide platforms. This is the only available preprocessing algorithm that can learn probe-level parameters based on sequential hyperparameter updates at small, consecutive batches of data, thus circumventing the extensive memory requirements of the standard approaches and opening up novel opportunities to take full advantage of contemporary microarray data collections. Moreover, using the most comprehensive data collections to estimate probe-level effects can assist in pinpointing individual probes affected by various biases and provide new tools to guide array design and quality control. The implementation is freely available in R/Bioconductor at http://www.bioconductor.org/packages/devel/bioc/html/RPA.html
* 20 pages, 3 figures, 1 supplementary PDF