 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeModelling-based experiment retrieval: A case study with gene expression clustering
Jan 04, 2016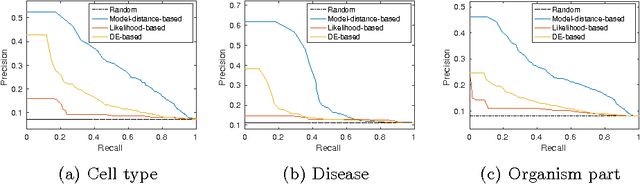
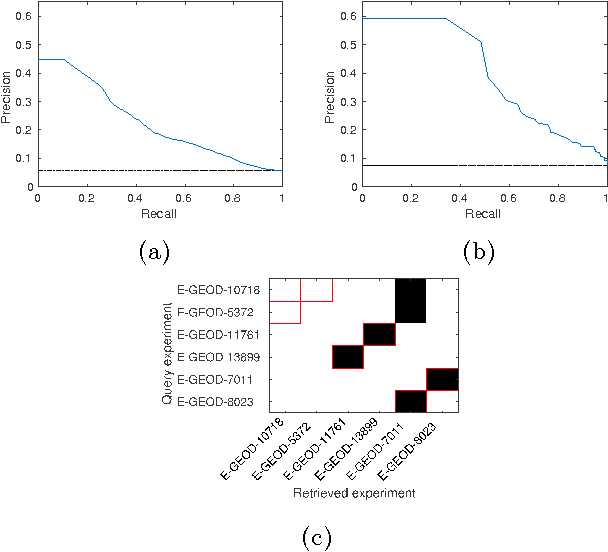
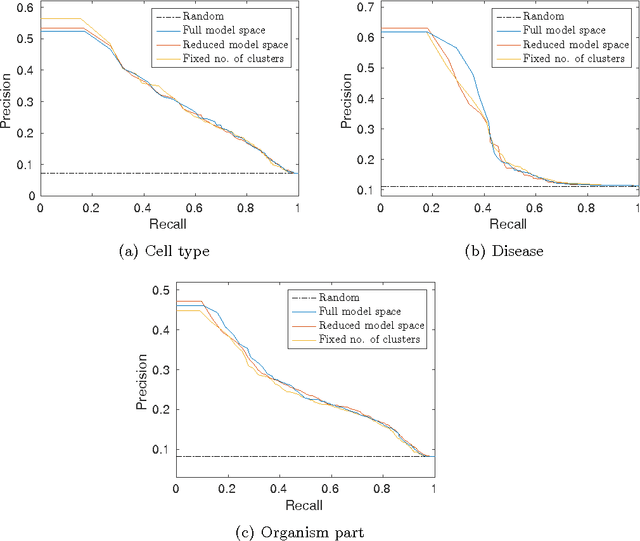
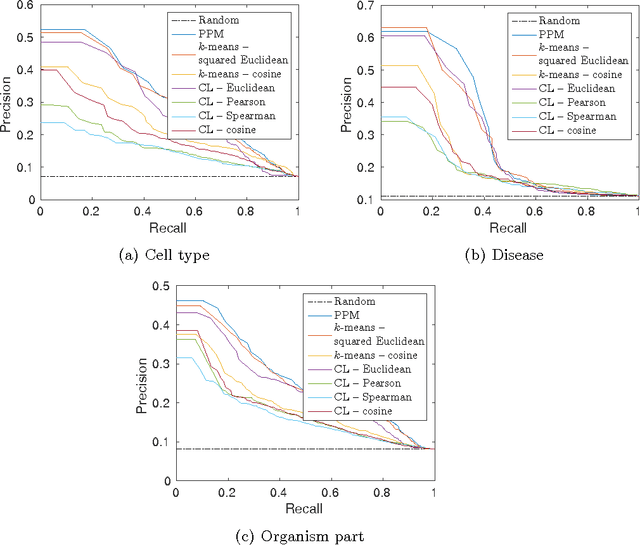
Motivation: Public and private repositories of experimental data are growing to sizes that require dedicated methods for finding relevant data. To improve on the state of the art of keyword searches from annotations, methods for content-based retrieval have been proposed. In the context of gene expression experiments, most methods retrieve gene expression profiles, requiring each experiment to be expressed as a single profile, typically of case vs. control. A more general, recently suggested alternative is to retrieve experiments whose models are good for modelling the query dataset. However, for very noisy and high-dimensional query data, this retrieval criterion turns out to be very noisy as well. Results: We propose doing retrieval using a denoised model of the query dataset, instead of the original noisy dataset itself. To this end, we introduce a general probabilistic framework, where each experiment is modelled separately and the retrieval is done by finding related models. For retrieval of gene expression experiments, we use a probabilistic model called product partition model, which induces a clustering of genes that show similar expression patterns across a number of samples. The suggested metric for retrieval using clusterings is the normalized information distance. Empirical results finally suggest that inference for the full probabilistic model can be approximated with good performance using computationally faster heuristic clustering approaches (e.g. $k$-means). The method is highly scalable and straightforward to apply to construct a general-purpose gene expression experiment retrieval method. Availability: The method can be implemented using standard clustering algorithms and normalized information distance, available in many statistical software packages.
Fully scalable online-preprocessing algorithm for short oligonucleotide microarray atlases
Dec 27, 2012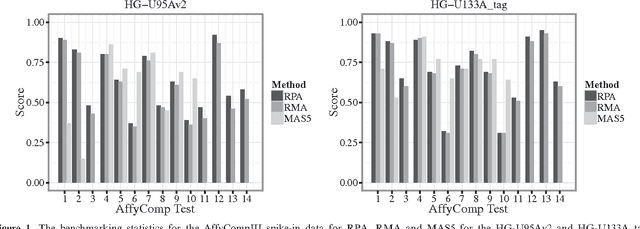
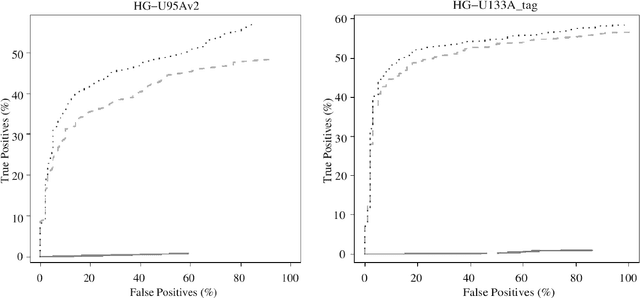
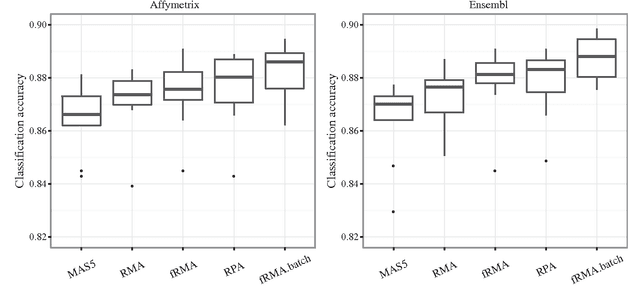
Accumulation of standardized data collections is opening up novel opportunities for holistic characterization of genome function. The limited scalability of current preprocessing techniques has, however, formed a bottleneck for full utilization of contemporary microarray collections. While short oligonucleotide arrays constitute a major source of genome-wide profiling data, scalable probe-level preprocessing algorithms have been available only for few measurement platforms based on pre-calculated model parameters from restricted reference training sets. To overcome these key limitations, we introduce a fully scalable online-learning algorithm that provides tools to process large microarray atlases including tens of thousands of arrays. Unlike the alternatives, the proposed algorithm scales up in linear time with respect to sample size and is readily applicable to all short oligonucleotide platforms. This is the only available preprocessing algorithm that can learn probe-level parameters based on sequential hyperparameter updates at small, consecutive batches of data, thus circumventing the extensive memory requirements of the standard approaches and opening up novel opportunities to take full advantage of contemporary microarray data collections. Moreover, using the most comprehensive data collections to estimate probe-level effects can assist in pinpointing individual probes affected by various biases and provide new tools to guide array design and quality control. The implementation is freely available in R/Bioconductor at http://www.bioconductor.org/packages/devel/bioc/html/RPA.html
* 20 pages, 3 figures, 1 supplementary PDF