 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBenchmarking community drug response prediction models: datasets, models, tools, and metrics for cross-dataset generalization analysis
Mar 18, 2025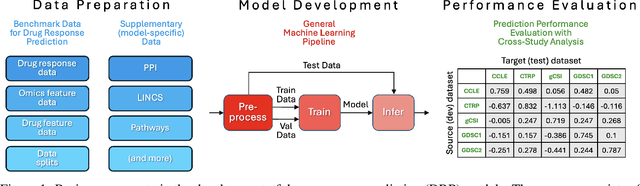
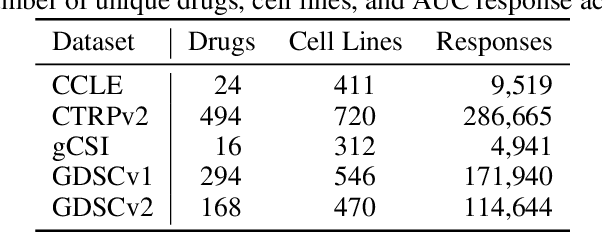
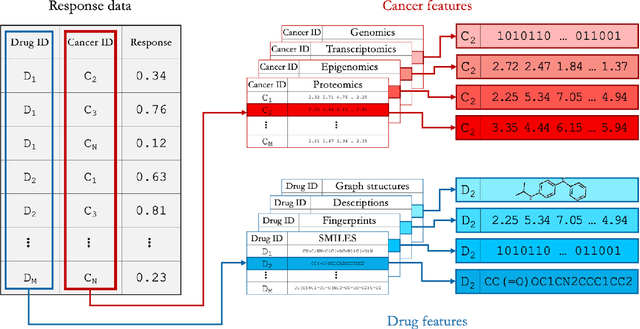

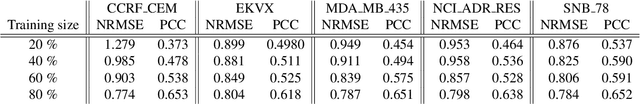
Deep learning (DL) and machine learning (ML) models have shown promise in drug response prediction (DRP), yet their ability to generalize across datasets remains an open question, raising concerns about their real-world applicability. Due to the lack of standardized benchmarking approaches, model evaluations and comparisons often rely on inconsistent datasets and evaluation criteria, making it difficult to assess true predictive capabilities. In this work, we introduce a benchmarking framework for evaluating cross-dataset prediction generalization in DRP models. Our framework incorporates five publicly available drug screening datasets, six standardized DRP models, and a scalable workflow for systematic evaluation. To assess model generalization, we introduce a set of evaluation metrics that quantify both absolute performance (e.g., predictive accuracy across datasets) and relative performance (e.g., performance drop compared to within-dataset results), enabling a more comprehensive assessment of model transferability. Our results reveal substantial performance drops when models are tested on unseen datasets, underscoring the importance of rigorous generalization assessments. While several models demonstrate relatively strong cross-dataset generalization, no single model consistently outperforms across all datasets. Furthermore, we identify CTRPv2 as the most effective source dataset for training, yielding higher generalization scores across target datasets. By sharing this standardized evaluation framework with the community, our study aims to establish a rigorous foundation for model comparison, and accelerate the development of robust DRP models for real-world applications.
Efficient Normalized Conformal Prediction and Uncertainty Quantification for Anti-Cancer Drug Sensitivity Prediction with Deep Regression Forests
Feb 21, 2024Deep learning models are being adopted and applied on various critical decision-making tasks, yet they are trained to provide point predictions without providing degrees of confidence. The trustworthiness of deep learning models can be increased if paired with uncertainty estimations. Conformal Prediction has emerged as a promising method to pair machine learning models with prediction intervals, allowing for a view of the model's uncertainty. However, popular uncertainty estimation methods for conformal prediction fail to provide heteroskedastic intervals that are equally accurate for all samples. In this paper, we propose a method to estimate the uncertainty of each sample by calculating the variance obtained from a Deep Regression Forest. We show that the deep regression forest variance improves the efficiency and coverage of normalized inductive conformal prediction on a drug response prediction task.
A Matrix Ensemble Kalman Filter-based Multi-arm Neural Network to Adequately Approximate Deep Neural Networks
Jul 19, 2023Deep Learners (DLs) are the state-of-art predictive mechanism with applications in many fields requiring complex high dimensional data processing. Although conventional DLs get trained via gradient descent with back-propagation, Kalman Filter (KF)-based techniques that do not need gradient computation have been developed to approximate DLs. We propose a multi-arm extension of a KF-based DL approximator that can mimic DL when the sample size is too small to train a multi-arm DL. The proposed Matrix Ensemble Kalman Filter-based multi-arm ANN (MEnKF-ANN) also performs explicit model stacking that becomes relevant when the training sample has an unequal-size feature set. Our proposed technique can approximate Long Short-term Memory (LSTM) Networks and attach uncertainty to the predictions obtained from these LSTMs with desirable coverage. We demonstrate how MEnKF-ANN can "adequately" approximate an LSTM network trained to classify what carbohydrate substrates are digested and utilized by a microbiome sample whose genomic sequences consist of polysaccharide utilization loci (PULs) and their encoded genes.
Investigation of REFINED CNN ensemble learning for anti-cancer drug sensitivity prediction
Sep 09, 2020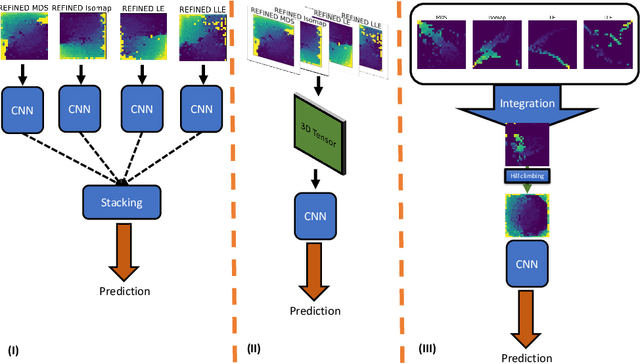
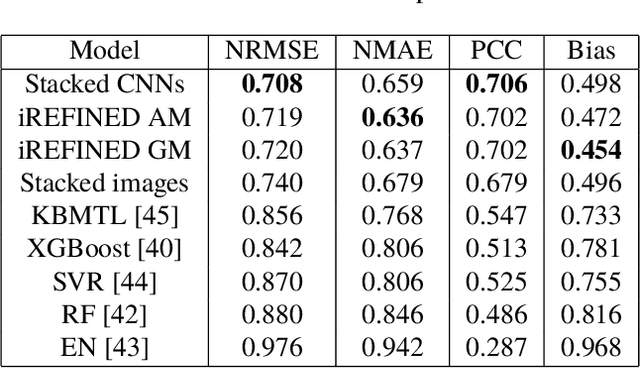

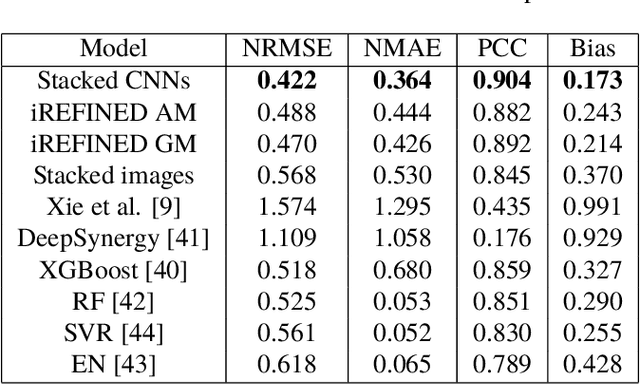
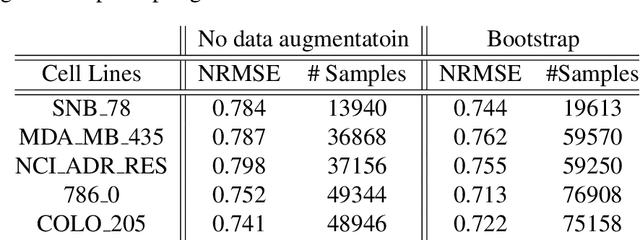
Anti-cancer drug sensitivity prediction using deep learning models for individual cell line is a significant challenge in personalized medicine. REFINED (REpresentation of Features as Images with NEighborhood Dependencies) CNN (Convolutional Neural Network) based models have shown promising results in drug sensitivity prediction. The primary idea behind REFINED CNN is representing high dimensional vectors as compact images with spatial correlations that can benefit from convolutional neural network architectures. However, the mapping from a vector to a compact 2D image is not unique due to variations in considered distance measures and neighborhoods. In this article, we consider predictions based on ensembles built from such mappings that can improve upon the best single REFINED CNN model prediction. Results illustrated using NCI60 and NCIALMANAC databases shows that the ensemble approaches can provide significant performance improvement as compared to individual models. We further illustrate that a single mapping created from the amalgamation of the different mappings can provide performance similar to stacking ensemble but with significantly lower computational complexity.
REFINED (REpresentation of Features as Images with NEighborhood Dependencies): A novel feature representation for Convolutional Neural Networks
Dec 11, 2019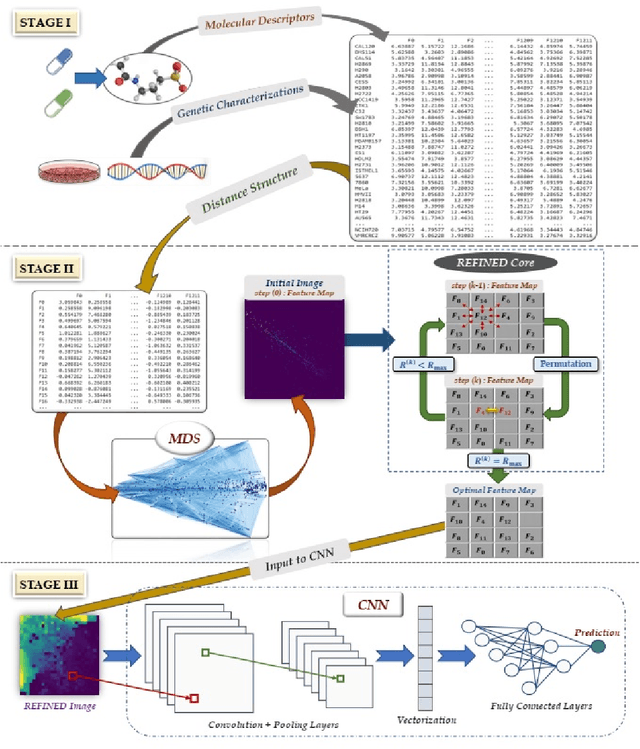
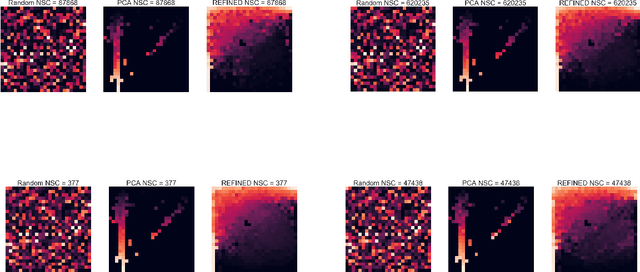
Deep learning with Convolutional Neural Networks has shown great promise in various areas of image-based classification and enhancement but is often unsuitable for predictive modeling involving non-image based features or features without spatial correlations. We present a novel approach for representation of high dimensional feature vector in a compact image form, termed REFINED (REpresentation of Features as Images with NEighborhood Dependencies), that is conducible for convolutional neural network based deep learning. We consider the correlations between features to generate a compact representation of the features in the form of a two-dimensional image using minimization of pairwise distances similar to multi-dimensional scaling. We hypothesize that this approach enables embedded feature selection and integrated with Convolutional Neural Network based Deep Learning can produce more accurate predictions as compared to Artificial Neural Networks, Random Forests and Support Vector Regression. We illustrate the superior predictive performance of the proposed representation, as compared to existing approaches, using synthetic datasets, cell line efficacy prediction based on drug chemical descriptors for NCI60 dataset and drug sensitivity prediction based on transcriptomic data and chemical descriptors using GDSC dataset. Results illustrated on both synthetic and biological datasets shows the higher prediction accuracy of the proposed framework as compared to existing methodologies while maintaining desirable properties in terms of bias and feature extraction.