 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSimCD: Simultaneous Clustering and Differential expression analysis for single-cell transcriptomic data
Apr 04, 2021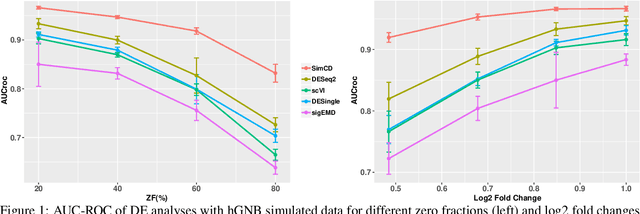
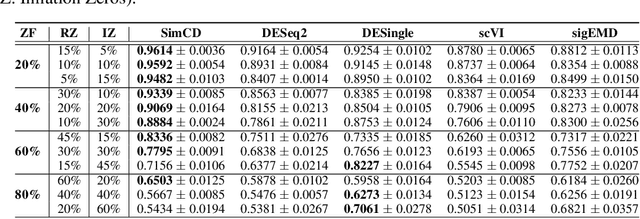
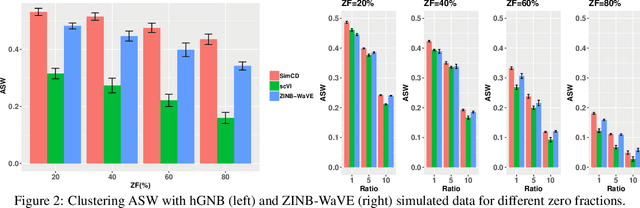
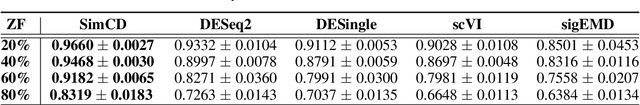
Single-Cell RNA sequencing (scRNA-seq) measurements have facilitated genome-scale transcriptomic profiling of individual cells, with the hope of deconvolving cellular dynamic changes in corresponding cell sub-populations to better understand molecular mechanisms of different development processes. Several scRNA-seq analysis methods have been proposed to first identify cell sub-populations by clustering and then separately perform differential expression analysis to understand gene expression changes. Their corresponding statistical models and inference algorithms are often designed disjointly. We develop a new method -- SimCD -- that explicitly models cell heterogeneity and dynamic differential changes in one unified hierarchical gamma-negative binomial (hGNB) model, allowing simultaneous cell clustering and differential expression analysis for scRNA-seq data. Our method naturally defines cell heterogeneity by dynamic expression changes, which is expected to help achieve better performances on the two tasks compared to the existing methods that perform them separately. In addition, SimCD better models dropout (zero inflation) in scRNA-seq data by both cell- and gene-level factors and obviates the need for sophisticated pre-processing steps such as normalization, thanks to the direct modeling of scRNA-seq count data by the rigorous hGNB model with an efficient Gibbs sampling inference algorithm. Extensive comparisons with the state-of-the-art methods on both simulated and real-world scRNA-seq count data demonstrate the capability of SimCD to discover cell clusters and capture dynamic expression changes. Furthermore, SimCD helps identify several known genes affected by food deprivation in hypothalamic neuron cell subtypes as well as some new potential markers, suggesting the capability of SimCD for bio-marker discovery.
Pairwise Supervised Hashing with Bernoulli Variational Auto-Encoder and Self-Control Gradient Estimator
May 21, 2020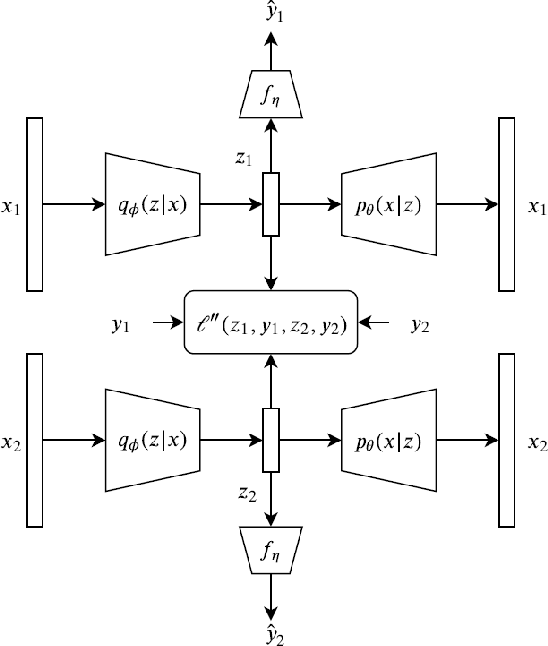
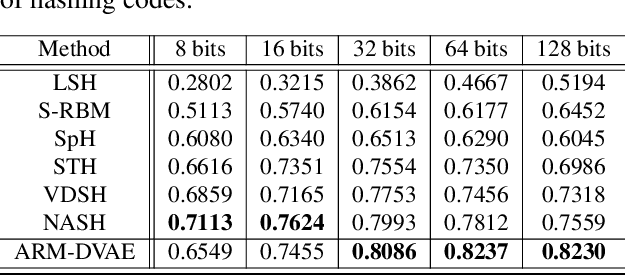
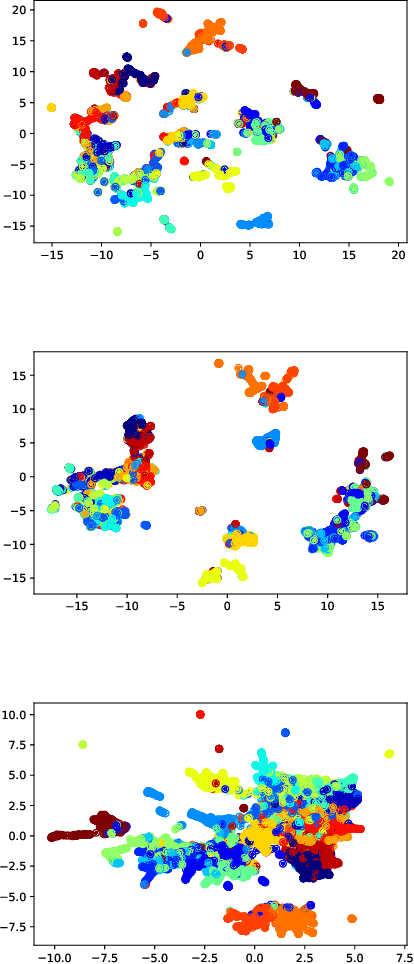
Semantic hashing has become a crucial component of fast similarity search in many large-scale information retrieval systems, in particular, for text data. Variational auto-encoders (VAEs) with binary latent variables as hashing codes provide state-of-the-art performance in terms of precision for document retrieval. We propose a pairwise loss function with discrete latent VAE to reward within-class similarity and between-class dissimilarity for supervised hashing. Instead of solving the optimization relying on existing biased gradient estimators, an unbiased low-variance gradient estimator is adopted to optimize the hashing function by evaluating the non-differentiable loss function over two correlated sets of binary hashing codes to control the variance of gradient estimates. This new semantic hashing framework achieves superior performance compared to the state-of-the-arts, as demonstrated by our comprehensive experiments.
* To appear in UAI 2020
Learnable Bernoulli Dropout for Bayesian Deep Learning
Feb 12, 2020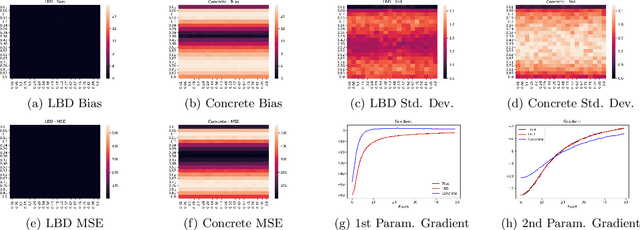
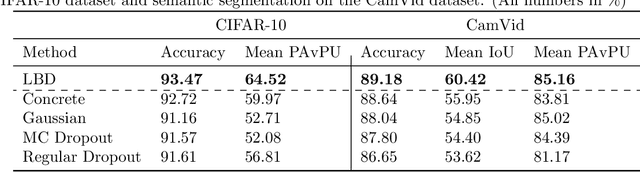
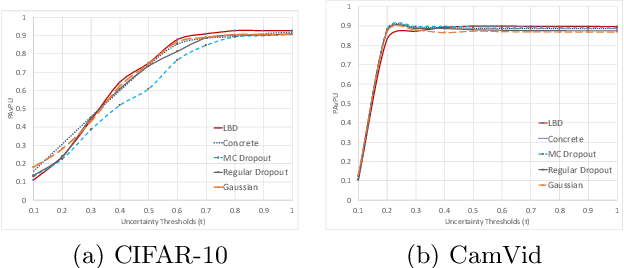
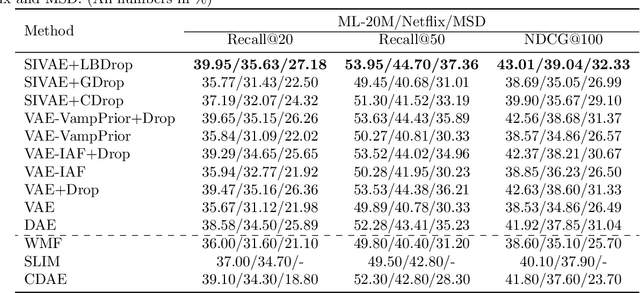
In this work, we propose learnable Bernoulli dropout (LBD), a new model-agnostic dropout scheme that considers the dropout rates as parameters jointly optimized with other model parameters. By probabilistic modeling of Bernoulli dropout, our method enables more robust prediction and uncertainty quantification in deep models. Especially, when combined with variational auto-encoders (VAEs), LBD enables flexible semi-implicit posterior representations, leading to new semi-implicit VAE~(SIVAE) models. We solve the optimization for training with respect to the dropout parameters using Augment-REINFORCE-Merge (ARM), an unbiased and low-variance gradient estimator. Our experiments on a range of tasks show the superior performance of our approach compared with other commonly used dropout schemes. Overall, LBD leads to improved accuracy and uncertainty estimates in image classification and semantic segmentation. Moreover, using SIVAE, we can achieve state-of-the-art performance on collaborative filtering for implicit feedback on several public datasets.
ARSM Gradient Estimator for Supervised Learning to Rank
Nov 01, 2019
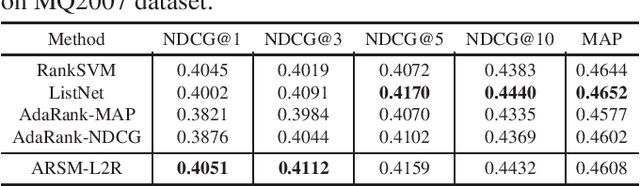
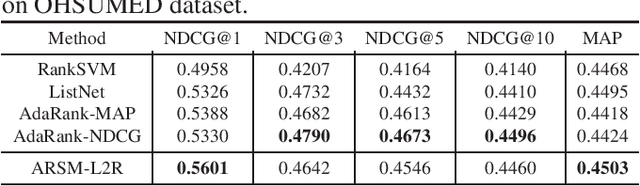
We propose a new model for supervised learning to rank. In our model, the relevancy labels are are assumed to follow a categorical distribution whose probabilities are constructed based on a scoring function. We optimize the training objective with respect to the multivariate categorical variables with an unbiased and low-variance gradient estimator. Learning to rank methods can generally be categorized into pointwise, pairwise, and listwise approaches. Our approach belongs to the class of pointwise methods. Although it has previously been reported that pointwise methods cannot achieve as good performance as of pairwise or listwise approaches, we show that the proposed method achieves better or comparable results on two datasets compared with pairwise and listwise methods.
Optimal Clustering with Missing Values
Feb 26, 2019
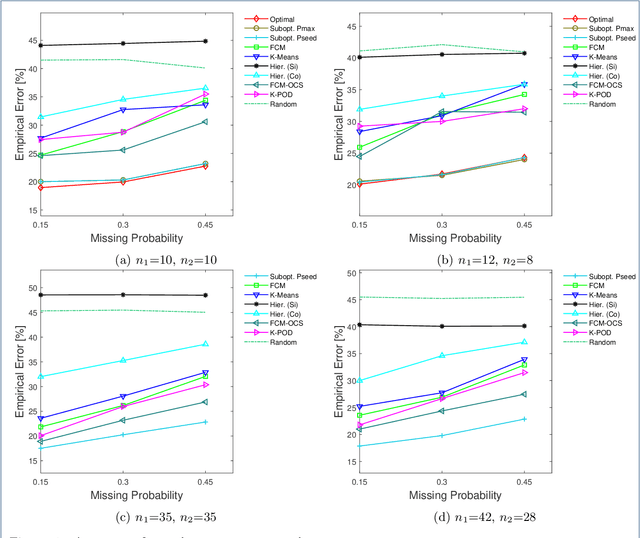
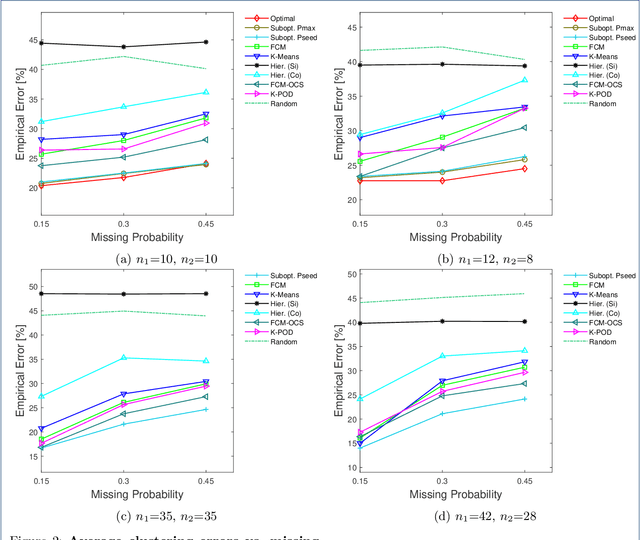
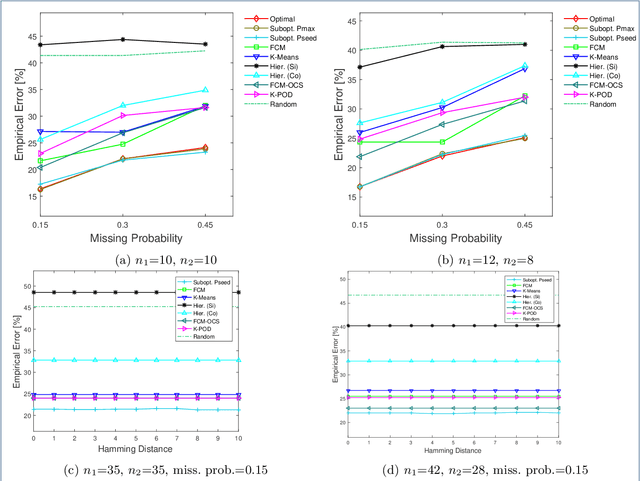
Missing values frequently arise in modern biomedical studies due to various reasons, including missing tests or complex profiling technologies for different omics measurements. Missing values can complicate the application of clustering algorithms, whose goals are to group points based on some similarity criterion. A common practice for dealing with missing values in the context of clustering is to first impute the missing values, and then apply the clustering algorithm on the completed data. We consider missing values in the context of optimal clustering, which finds an optimal clustering operator with reference to an underlying random labeled point process (RLPP). We show how the missing-value problem fits neatly into the overall framework of optimal clustering by incorporating the missing value mechanism into the random labeled point process and then marginalizing out the missing-value process. In particular, we demonstrate the proposed framework for the Gaussian model with arbitrary covariance structures. Comprehensive experimental studies on both synthetic and real-world RNA-seq data show the superior performance of the proposed optimal clustering with missing values when compared to various clustering approaches. Optimal clustering with missing values obviates the need for imputation-based pre-processing of the data, while at the same time possessing smaller clustering errors.
Bayesian multi-domain learning for cancer subtype discovery from next-generation sequencing count data
Oct 22, 2018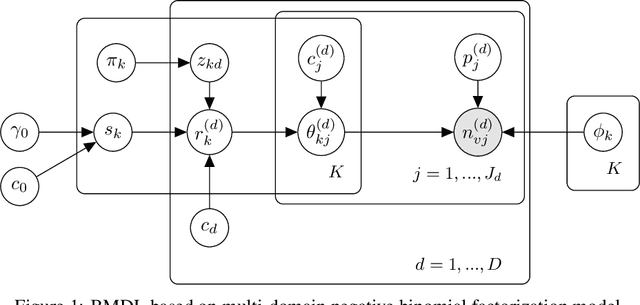
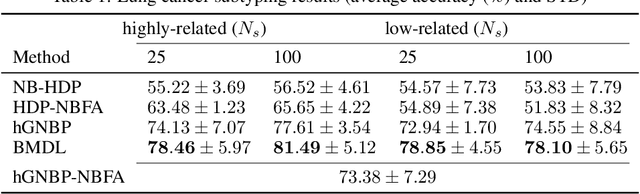
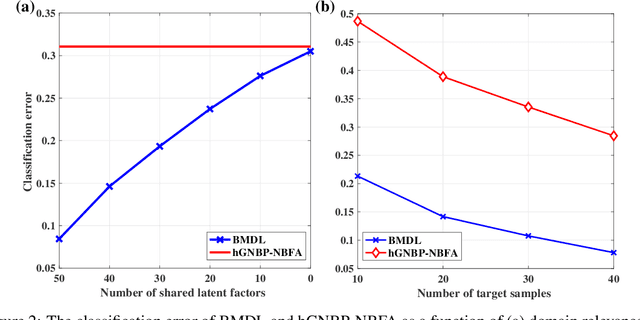
Precision medicine aims for personalized prognosis and therapeutics by utilizing recent genome-scale high-throughput profiling techniques, including next-generation sequencing (NGS). However, translating NGS data faces several challenges. First, NGS count data are often overdispersed, requiring appropriate modeling. Second, compared to the number of involved molecules and system complexity, the number of available samples for studying complex disease, such as cancer, is often limited, especially considering disease heterogeneity. The key question is whether we may integrate available data from all different sources or domains to achieve reproducible disease prognosis based on NGS count data. In this paper, we develop a Bayesian Multi-Domain Learning (BMDL) model that derives domain-dependent latent representations of overdispersed count data based on hierarchical negative binomial factorization for accurate cancer subtyping even if the number of samples for a specific cancer type is small. Experimental results from both our simulated and NGS datasets from The Cancer Genome Atlas (TCGA) demonstrate the promising potential of BMDL for effective multi-domain learning without "negative transfer" effects often seen in existing multi-task learning and transfer learning methods.
Differential Expression Analysis of Dynamical Sequencing Count Data with a Gamma Markov Chain
Mar 07, 2018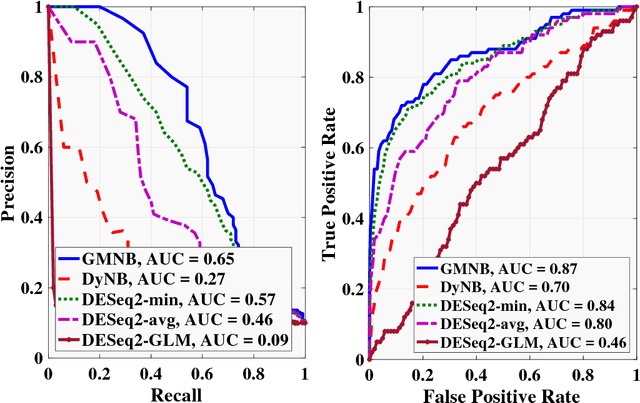
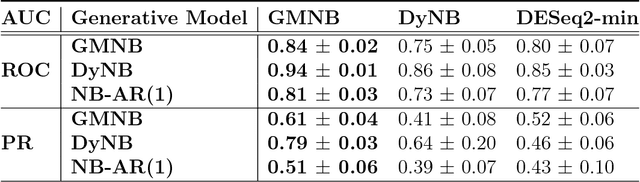
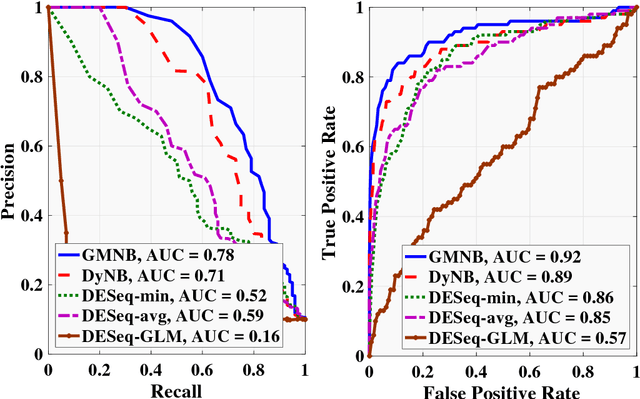
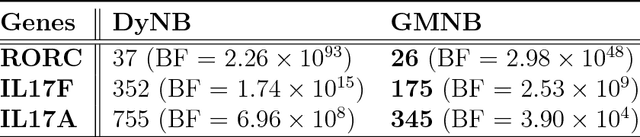
Next-generation sequencing (NGS) to profile temporal changes in living systems is gaining more attention for deriving better insights into the underlying biological mechanisms compared to traditional static sequencing experiments. Nonetheless, the majority of existing statistical tools for analyzing NGS data lack the capability of exploiting the richer information embedded in temporal data. Several recent tools have been developed to analyze such data but they typically impose strict model assumptions, such as smoothness on gene expression dynamic changes. To capture a broader range of gene expression dynamic patterns, we develop the gamma Markov negative binomial (GMNB) model that integrates a gamma Markov chain into a negative binomial distribution model, allowing flexible temporal variation in NGS count data. Using Bayes factors, GMNB enables more powerful temporal gene differential expression analysis across different phenotypes or treatment conditions. In addition, it naturally handles the heterogeneity of sequencing depth in different samples, removing the need for ad-hoc normalization. Efficient Gibbs sampling inference of the GMNB model parameters is achieved by exploiting novel data augmentation techniques. Extensive experiments on both simulated and real-world RNA-seq data show that GMNB outperforms existing methods in both receiver operating characteristic (ROC) and precision-recall (PR) curves of differential expression analysis results.