 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLoRA-BERT: a Natural Language Processing Model for Robust and Accurate Prediction of long non-coding RNAs
Nov 11, 2024Long non-coding RNAs (lncRNAs) serve as crucial regulators in numerous biological processes. Although they share sequence similarities with messenger RNAs (mRNAs), lncRNAs perform entirely different roles, providing new avenues for biological research. The emergence of next-generation sequencing technologies has greatly advanced the detection and identification of lncRNA transcripts and deep learning-based approaches have been introduced to classify long non-coding RNAs (lncRNAs). These advanced methods have significantly enhanced the efficiency of identifying lncRNAs. However, many of these methods are devoid of robustness and accuracy due to the extended length of the sequences involved. To tackle this issue, we have introduced a novel pre-trained bidirectional encoder representation called LoRA-BERT. LoRA-BERT is designed to capture the importance of nucleotide-level information during sequence classification, leading to more robust and satisfactory outcomes. In a comprehensive comparison with commonly used sequence prediction tools, we have demonstrated that LoRA-BERT outperforms them in terms of accuracy and efficiency. Our results indicate that, when utilizing the transformer model, LoRA-BERT achieves state-of-the-art performance in predicting both lncRNAs and mRNAs for human and mouse species. Through the utilization of LoRA-BERT, we acquire valuable insights into the traits of lncRNAs and mRNAs, offering the potential to aid in the comprehension and detection of diseases linked to lncRNAs in humans.
Differential Expression Analysis of Dynamical Sequencing Count Data with a Gamma Markov Chain
Mar 07, 2018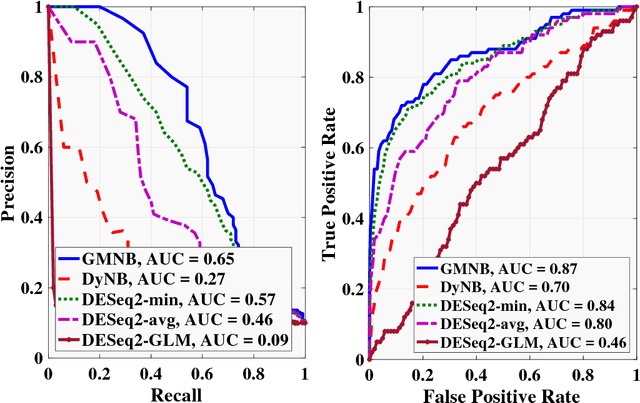
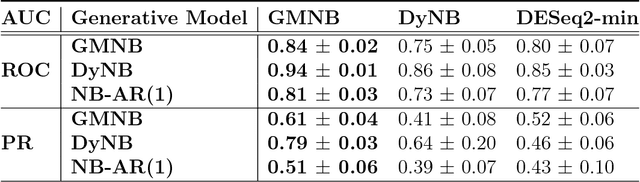
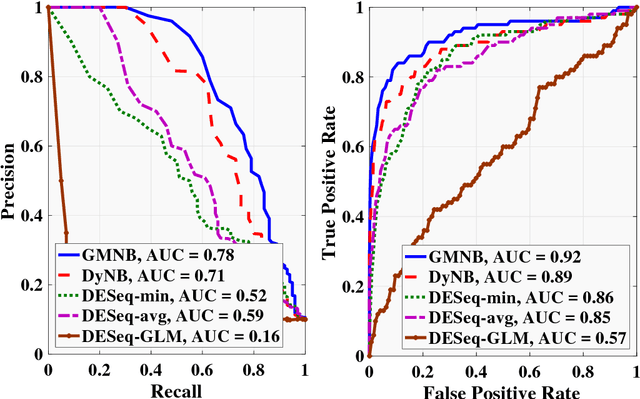
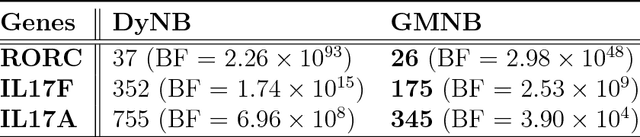
Next-generation sequencing (NGS) to profile temporal changes in living systems is gaining more attention for deriving better insights into the underlying biological mechanisms compared to traditional static sequencing experiments. Nonetheless, the majority of existing statistical tools for analyzing NGS data lack the capability of exploiting the richer information embedded in temporal data. Several recent tools have been developed to analyze such data but they typically impose strict model assumptions, such as smoothness on gene expression dynamic changes. To capture a broader range of gene expression dynamic patterns, we develop the gamma Markov negative binomial (GMNB) model that integrates a gamma Markov chain into a negative binomial distribution model, allowing flexible temporal variation in NGS count data. Using Bayes factors, GMNB enables more powerful temporal gene differential expression analysis across different phenotypes or treatment conditions. In addition, it naturally handles the heterogeneity of sequencing depth in different samples, removing the need for ad-hoc normalization. Efficient Gibbs sampling inference of the GMNB model parameters is achieved by exploiting novel data augmentation techniques. Extensive experiments on both simulated and real-world RNA-seq data show that GMNB outperforms existing methods in both receiver operating characteristic (ROC) and precision-recall (PR) curves of differential expression analysis results.