 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCOVID-Datathon: Biomarker identification for COVID-19 severity based on BALF scRNA-seq data
Oct 11, 2021


The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emergence began in late 2019 and has since spread rapidly worldwide. The characteristics of respiratory immune response to this emerging virus is not clear. Recently, Single-cell RNA sequencing (scRNA-seq) transcriptome profiling of Bronchoalveolar lavage fluid (BALF) cells has been done to elucidate the potential mechanisms underlying in COVID-19. With the aim of better utilizing this atlas of BALF cells in response to the virus, here we propose a bioinformatics pipeline to identify candidate biomarkers of COVID-19 severity, which may help characterize BALF cells to have better mechanistic understanding of SARS-CoV-2 infection. The proposed pipeline is implemented in R and is available at https://github.com/namini94/scBALF_Hackathon.
SimCD: Simultaneous Clustering and Differential expression analysis for single-cell transcriptomic data
Apr 04, 2021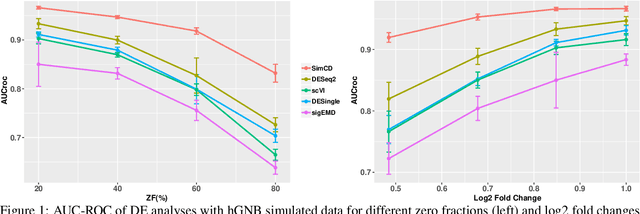
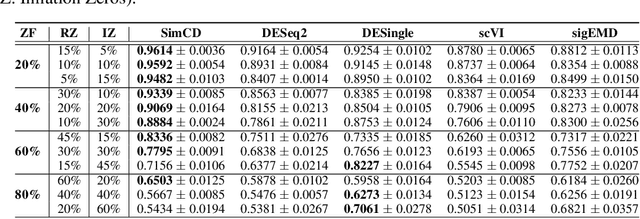
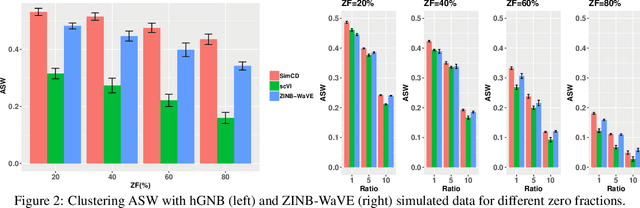
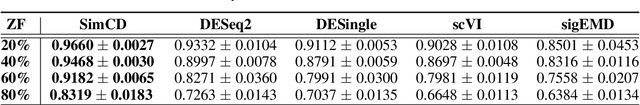
Single-Cell RNA sequencing (scRNA-seq) measurements have facilitated genome-scale transcriptomic profiling of individual cells, with the hope of deconvolving cellular dynamic changes in corresponding cell sub-populations to better understand molecular mechanisms of different development processes. Several scRNA-seq analysis methods have been proposed to first identify cell sub-populations by clustering and then separately perform differential expression analysis to understand gene expression changes. Their corresponding statistical models and inference algorithms are often designed disjointly. We develop a new method -- SimCD -- that explicitly models cell heterogeneity and dynamic differential changes in one unified hierarchical gamma-negative binomial (hGNB) model, allowing simultaneous cell clustering and differential expression analysis for scRNA-seq data. Our method naturally defines cell heterogeneity by dynamic expression changes, which is expected to help achieve better performances on the two tasks compared to the existing methods that perform them separately. In addition, SimCD better models dropout (zero inflation) in scRNA-seq data by both cell- and gene-level factors and obviates the need for sophisticated pre-processing steps such as normalization, thanks to the direct modeling of scRNA-seq count data by the rigorous hGNB model with an efficient Gibbs sampling inference algorithm. Extensive comparisons with the state-of-the-art methods on both simulated and real-world scRNA-seq count data demonstrate the capability of SimCD to discover cell clusters and capture dynamic expression changes. Furthermore, SimCD helps identify several known genes affected by food deprivation in hypothalamic neuron cell subtypes as well as some new potential markers, suggesting the capability of SimCD for bio-marker discovery.