 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024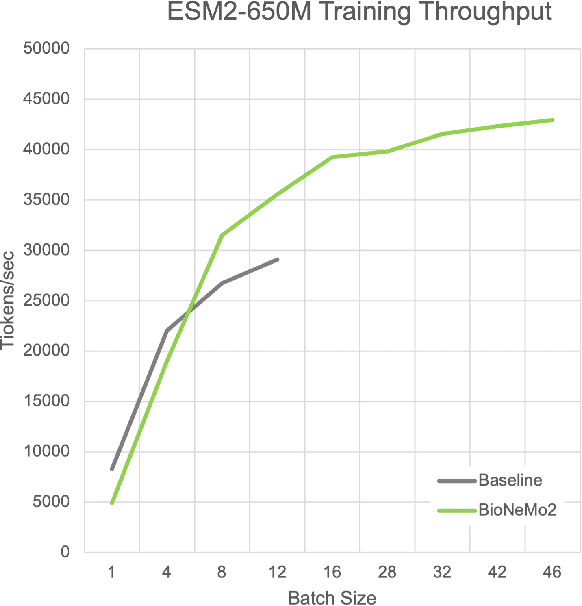
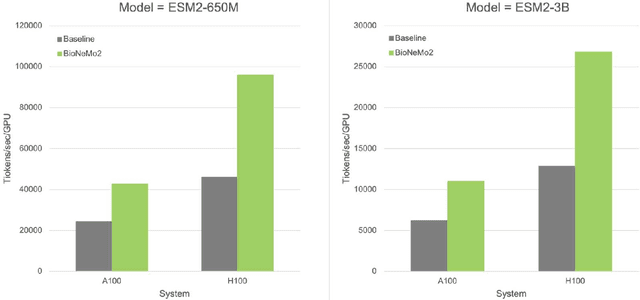
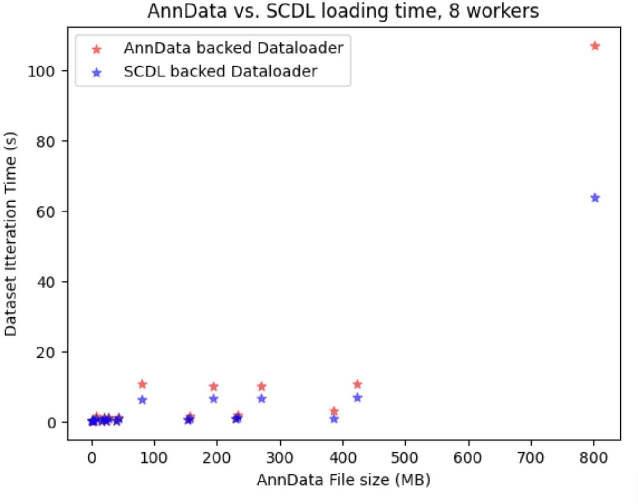
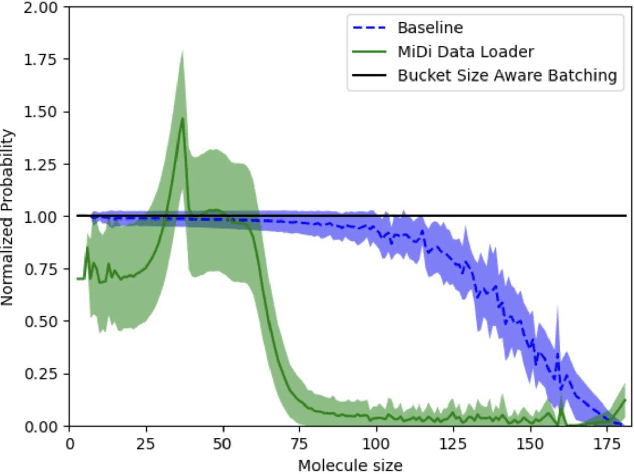
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
Beyond the training set: an intuitive method for detecting distribution shift in model-based optimization
Nov 09, 2023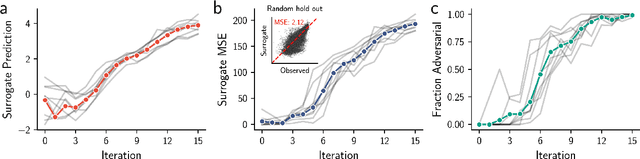
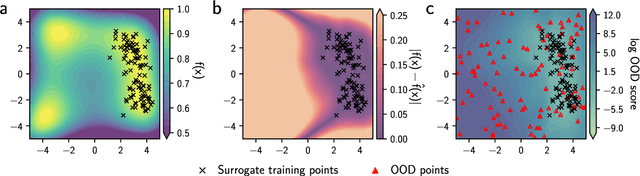
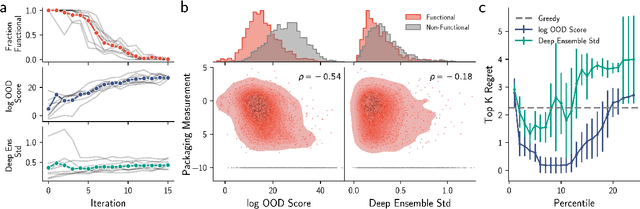
Model-based optimization (MBO) is increasingly applied to design problems in science and engineering. A common scenario involves using a fixed training set to train models, with the goal of designing new samples that outperform those present in the training data. A major challenge in this setting is distribution shift, where the distributions of training and design samples are different. While some shift is expected, as the goal is to create better designs, this change can negatively affect model accuracy and subsequently, design quality. Despite the widespread nature of this problem, addressing it demands deep domain knowledge and artful application. To tackle this issue, we propose a straightforward method for design practitioners that detects distribution shifts. This method trains a binary classifier using knowledge of the unlabeled design distribution to separate the training data from the design data. The classifier's logit scores are then used as a proxy measure of distribution shift. We validate our method in a real-world application by running offline MBO and evaluate the effect of distribution shift on design quality. We find that the intensity of the shift in the design distribution varies based on the number of steps taken by the optimization algorithm, and our simple approach can identify these shifts. This enables users to constrain their search to regions where the model's predictions are reliable, thereby increasing the quality of designs.
Contrastive losses as generalized models of global epistasis
May 08, 2023Fitness functions map large combinatorial spaces of biological sequences to properties of interest. Inferring these multimodal functions from experimental data is a central task in modern protein engineering. Global epistasis models are an effective and physically-grounded class of models for estimating fitness functions from observed data. These models assume that a sparse latent function is transformed by a monotonic nonlinearity to emit measurable fitness. Here we demonstrate that minimizing contrastive loss functions, such as the Bradley-Terry loss, is a simple and flexible technique for extracting the sparse latent function implied by global epistasis. We argue by way of a fitness-epistasis uncertainty principle that the nonlinearities in global epistasis models can produce observed fitness functions that do not admit sparse representations, and thus may be inefficient to learn from observations when using a Mean Squared Error (MSE) loss (a common practice). We show that contrastive losses are able to accurately estimate a ranking function from limited data even in regimes where MSE is ineffective. We validate the practical utility of this insight by showing contrastive loss functions result in consistently improved performance on benchmark tasks.
Forecasting labels under distribution-shift for machine-guided sequence design
Nov 18, 2022The ability to design and optimize biological sequences with specific functionalities would unlock enormous value in technology and healthcare. In recent years, machine learning-guided sequence design has progressed this goal significantly, though validating designed sequences in the lab or clinic takes many months and substantial labor. It is therefore valuable to assess the likelihood that a designed set contains sequences of the desired quality (which often lies outside the label distribution in our training data) before committing resources to an experiment. Forecasting, a prominent concept in many domains where feedback can be delayed (e.g. elections), has not been used or studied in the context of sequence design. Here we propose a method to guide decision-making that forecasts the performance of high-throughput libraries (e.g. containing $10^5$ unique variants) based on estimates provided by models, providing a posterior for the distribution of labels in the library. We show that our method outperforms baselines that naively use model scores to estimate library performance, which are the only tool available today for this purpose.
A primer on model-guided exploration of fitness landscapes for biological sequence design
Oct 23, 2020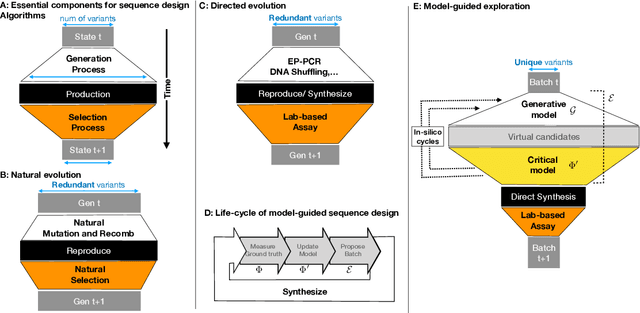
Machine learning methods are increasingly employed to address challenges faced by biologists. One area that will greatly benefit from this cross-pollination is the problem of biological sequence design, which has massive potential for therapeutic applications. However, significant inefficiencies remain in communication between these fields which result in biologists finding the progress in machine learning inaccessible, and hinder machine learning scientists from contributing to impactful problems in bioengineering. Sequence design can be seen as a search process on a discrete, high-dimensional space, where each sequence is associated with a function. This sequence-to-function map is known as a "Fitness Landscape". Designing a sequence with a particular function is hence a matter of "discovering" such a (often rare) sequence within this space. Today we can build predictive models with good interpolation ability due to impressive progress in the synthesis and testing of biological sequences in large numbers, which enables model training and validation. However, it often remains a challenge to find useful sequences with the properties that we like using these models. In particular, in this primer we highlight that algorithms for experimental design, what we call "exploration strategies", are a related, yet distinct problem from building good models of sequence-to-function maps. We review advances and insights from current literature -- by no means a complete treatment -- while highlighting desirable features of optimal model-guided exploration, and cover potential pitfalls drawn from our own experience. This primer can serve as a starting point for researchers from different domains that are interested in the problem of searching a sequence space with a model, but are perhaps unaware of approaches that originate outside their field.
AdaLead: A simple and robust adaptive greedy search algorithm for sequence design
Oct 05, 2020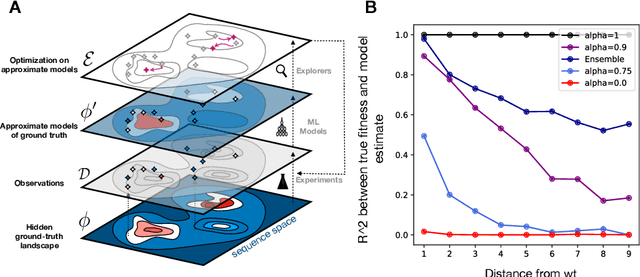
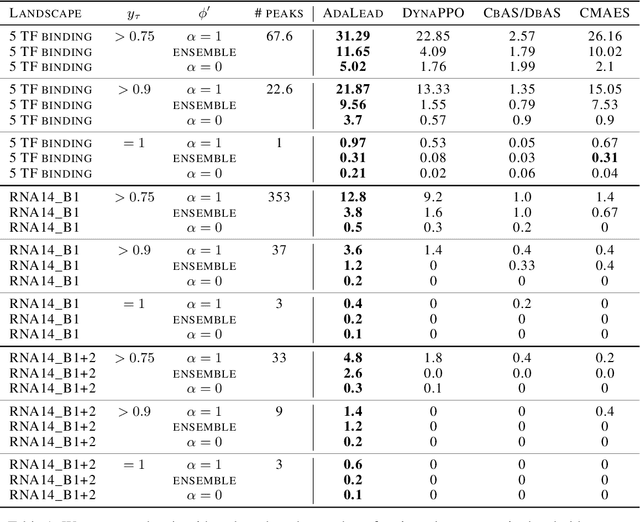
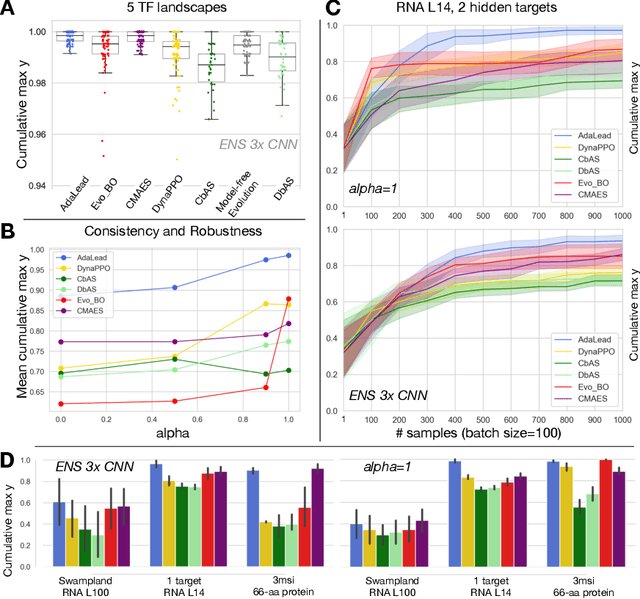
Efficient design of biological sequences will have a great impact across many industrial and healthcare domains. However, discovering improved sequences requires solving a difficult optimization problem. Traditionally, this challenge was approached by biologists through a model-free method known as "directed evolution", the iterative process of random mutation and selection. As the ability to build models that capture the sequence-to-function map improves, such models can be used as oracles to screen sequences before running experiments. In recent years, interest in better algorithms that effectively use such oracles to outperform model-free approaches has intensified. These span from approaches based on Bayesian Optimization, to regularized generative models and adaptations of reinforcement learning. In this work, we implement an open-source Fitness Landscape EXploration Sandbox (FLEXS: github.com/samsinai/FLEXS) environment to test and evaluate these algorithms based on their optimality, consistency, and robustness. Using FLEXS, we develop an easy-to-implement, scalable, and robust evolutionary greedy algorithm (AdaLead). Despite its simplicity, we show that AdaLead is a remarkably strong benchmark that out-competes more complex state of the art approaches in a variety of biologically motivated sequence design challenges.
Variational auto-encoding of protein sequences
Jan 03, 2018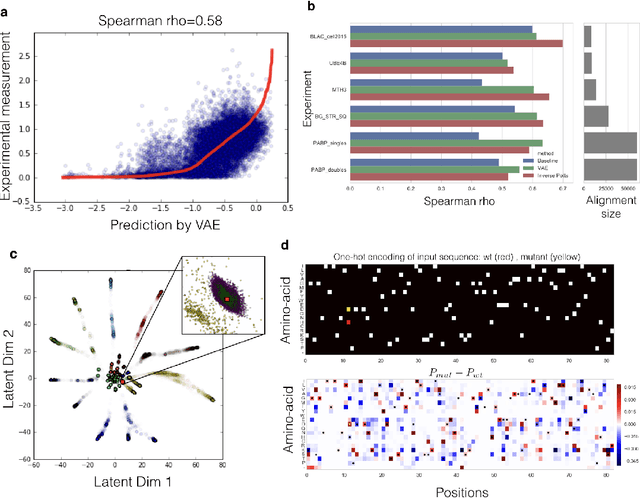
Proteins are responsible for the most diverse set of functions in biology. The ability to extract information from protein sequences and to predict the effects of mutations is extremely valuable in many domains of biology and medicine. However the mapping between protein sequence and function is complex and poorly understood. Here we present an embedding of natural protein sequences using a Variational Auto-Encoder and use it to predict how mutations affect protein function. We use this unsupervised approach to cluster natural variants and learn interactions between sets of positions within a protein. This approach generally performs better than baseline methods that consider no interactions within sequences, and in some cases better than the state-of-the-art approaches that use the inverse-Potts model. This generative model can be used to computationally guide exploration of protein sequence space and to better inform rational and automatic protein design.