 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeContrastive losses as generalized models of global epistasis
May 08, 2023Fitness functions map large combinatorial spaces of biological sequences to properties of interest. Inferring these multimodal functions from experimental data is a central task in modern protein engineering. Global epistasis models are an effective and physically-grounded class of models for estimating fitness functions from observed data. These models assume that a sparse latent function is transformed by a monotonic nonlinearity to emit measurable fitness. Here we demonstrate that minimizing contrastive loss functions, such as the Bradley-Terry loss, is a simple and flexible technique for extracting the sparse latent function implied by global epistasis. We argue by way of a fitness-epistasis uncertainty principle that the nonlinearities in global epistasis models can produce observed fitness functions that do not admit sparse representations, and thus may be inefficient to learn from observations when using a Mean Squared Error (MSE) loss (a common practice). We show that contrastive losses are able to accurately estimate a ranking function from limited data even in regimes where MSE is ineffective. We validate the practical utility of this insight by showing contrastive loss functions result in consistently improved performance on benchmark tasks.
Learning the shape of protein micro-environments with a holographic convolutional neural network
Nov 05, 2022Proteins play a central role in biology from immune recognition to brain activity. While major advances in machine learning have improved our ability to predict protein structure from sequence, determining protein function from structure remains a major challenge. Here, we introduce Holographic Convolutional Neural Network (H-CNN) for proteins, which is a physically motivated machine learning approach to model amino acid preferences in protein structures. H-CNN reflects physical interactions in a protein structure and recapitulates the functional information stored in evolutionary data. H-CNN accurately predicts the impact of mutations on protein function, including stability and binding of protein complexes. Our interpretable computational model for protein structure-function maps could guide design of novel proteins with desired function.
Quantitative genetic algorithms
Dec 06, 2019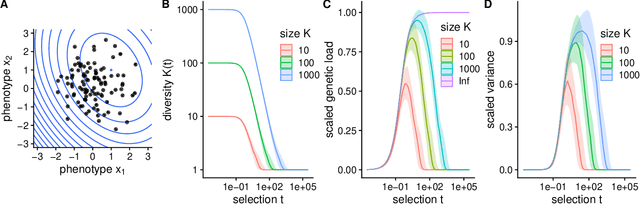
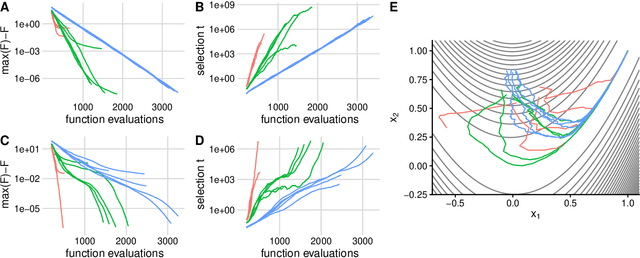
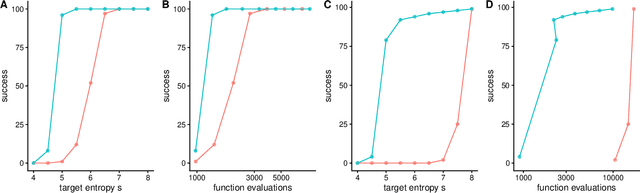
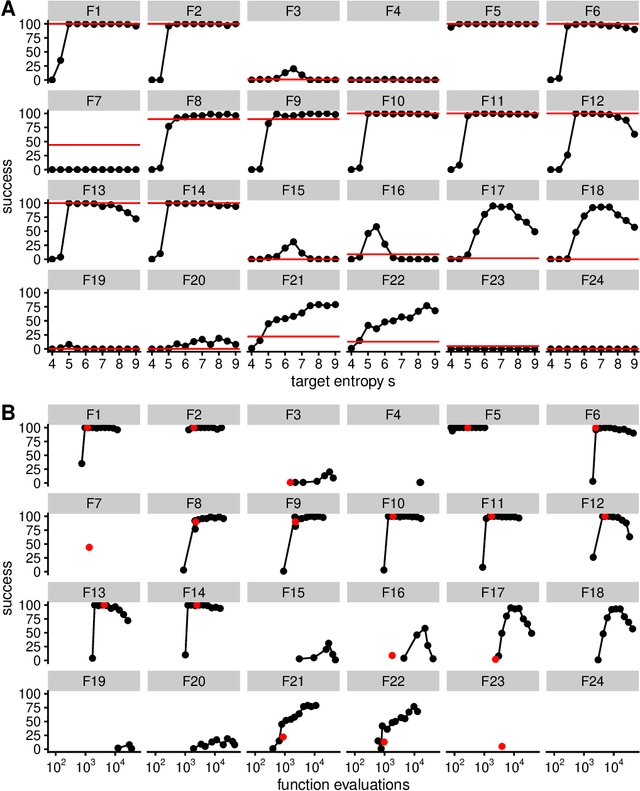
Evolutionary algorithms, inspired by natural evolution, aim to optimize difficult objective functions without computing derivatives. Here we detail the relationship between population genetics and evolutionary optimization and formulate a new evolutionary algorithm. Given a distribution of phenotypes on a fitness landscape, we summarize how natural selection moves a population in the direction of a natural gradient, similar to natural gradient descent, and show how intermediate selection is most informative of the fitness landscape. Then we describe the generation of new candidate solutions and propose an operator that recombines the whole population to generate variants that preserve normal statistics. Finally we combine natural selection, our recombination operator, and an adaptive method to increase selection to create a quantitative genetic algorithm (QGA). QGA is similar to covariance matrix adaptation and natural evolutionary strategies in optimization, with similar performance, although QGA requires tuning of its single hyperparameter. QGA is extremely simple in implementation with no matrix inversion or factorization, does not require storing a covariance matrix, is trivial to parallelize, and may form the basis of more robust algorithms.