 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCompressed representation of brain genetic transcription
Oct 24, 2023The architecture of the brain is too complex to be intuitively surveyable without the use of compressed representations that project its variation into a compact, navigable space. The task is especially challenging with high-dimensional data, such as gene expression, where the joint complexity of anatomical and transcriptional patterns demands maximum compression. Established practice is to use standard principal component analysis (PCA), whose computational felicity is offset by limited expressivity, especially at great compression ratios. Employing whole-brain, voxel-wise Allen Brain Atlas transcription data, here we systematically compare compressed representations based on the most widely supported linear and non-linear methods-PCA, kernel PCA, non-negative matrix factorization (NMF), t-stochastic neighbour embedding (t-SNE), uniform manifold approximation and projection (UMAP), and deep auto-encoding-quantifying reconstruction fidelity, anatomical coherence, and predictive utility with respect to signalling, microstructural, and metabolic targets. We show that deep auto-encoders yield superior representations across all metrics of performance and target domains, supporting their use as the reference standard for representing transcription patterns in the human brain.
The legibility of the imaged human brain
Aug 23, 2023

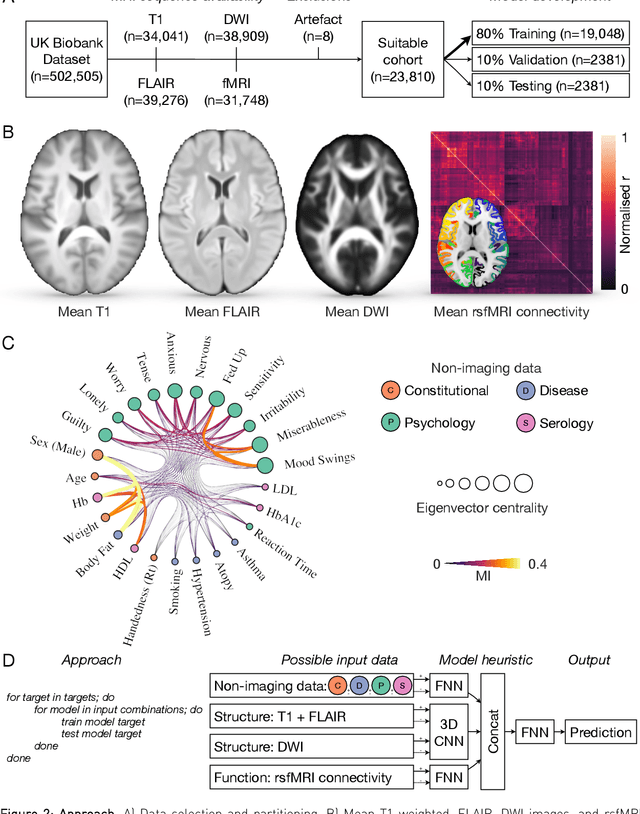

Our knowledge of the organisation of the human brain at the population-level is yet to translate into power to predict functional differences at the individual-level, limiting clinical applications, and casting doubt on the generalisability of inferred mechanisms. It remains unknown whether the difficulty arises from the absence of individuating biological patterns within the brain, or from limited power to access them with the models and compute at our disposal. Here we comprehensively investigate the resolvability of such patterns with data and compute at unprecedented scale. Across 23810 unique participants from UK Biobank, we systematically evaluate the predictability of 25 individual biological characteristics, from all available combinations of structural and functional neuroimaging data. Over 4526 GPU*hours of computation, we train, optimize, and evaluate out-of-sample 700 individual predictive models, including multilayer perceptrons of demographic, psychological, serological, chronic morbidity, and functional connectivity characteristics, and both uni- and multi-modal 3D convolutional neural network models of macro- and micro-structural brain imaging. We find a marked discrepancy between the high predictability of sex (balanced accuracy 99.7%), age (mean absolute error 2.048 years, R2 0.859), and weight (mean absolute error 2.609Kg, R2 0.625), for which we set new state-of-the-art performance, and the surprisingly low predictability of other characteristics. Neither structural nor functional imaging predicted individual psychology better than the coincidence of common chronic morbidity (p<0.05). Serology predicted common morbidity (p<0.05) and was best predicted by it (p<0.001), followed by structural neuroimaging (p<0.05). Our findings suggest either more informative imaging or more powerful models will be needed to decipher individual level characteristics from the brain.
The minimal computational substrate of fluid intelligence
Aug 14, 2023The quantification of cognitive powers rests on identifying a behavioural task that depends on them. Such dependence cannot be assured, for the powers a task invokes cannot be experimentally controlled or constrained a priori, resulting in unknown vulnerability to failure of specificity and generalisability. Evaluating a compact version of Raven's Advanced Progressive Matrices (RAPM), a widely used clinical test of fluid intelligence, we show that LaMa, a self-supervised artificial neural network trained solely on the completion of partially masked images of natural environmental scenes, achieves human-level test scores a prima vista, without any task-specific inductive bias or training. Compared with cohorts of healthy and focally lesioned participants, LaMa exhibits human-like variation with item difficulty, and produces errors characteristic of right frontal lobe damage under degradation of its ability to integrate global spatial patterns. LaMa's narrow training and limited capacity -- comparable to the nervous system of the fruit fly -- suggest RAPM may be open to computationally simple solutions that need not necessarily invoke abstract reasoning.
Translating automated brain tumour phenotyping to clinical neuroimaging
Jun 13, 2022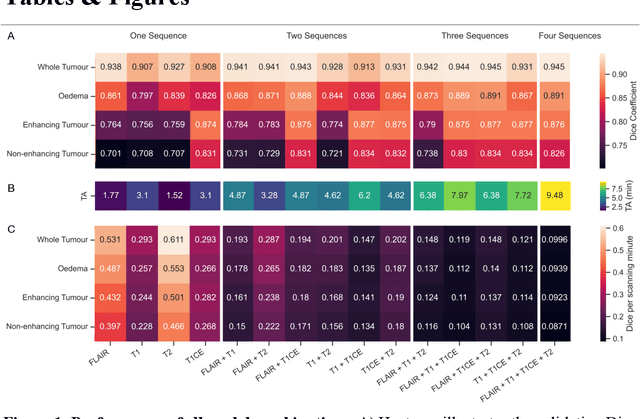
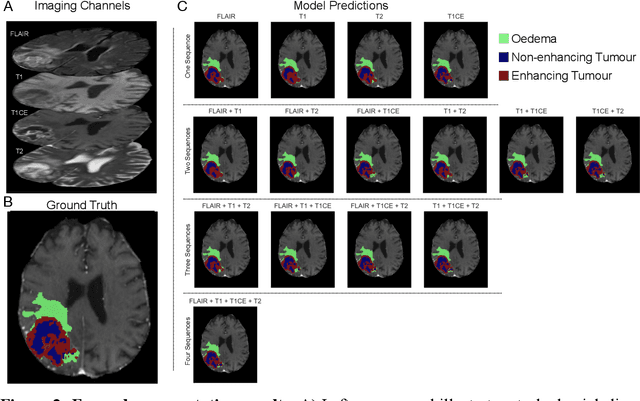


Background: The complex heterogeneity of brain tumours is increasingly recognized to demand data of magnitudes and richness only fully-inclusive, large-scale collections drawn from routine clinical care could plausibly offer. This is a task contemporary machine learning could facilitate, especially in neuroimaging, but its ability to deal with incomplete data common in real world clinical practice remains unknown. Here we apply state-of-the-art methods to large scale, multi-site MRI data to quantify the comparative fidelity of automated tumour segmentation models replicating the various levels of completeness observed in clinical reality. Methods: We compare deep learning (nnU-Net-derived) tumour segmentation models with all possible combinations of T1, contrast-enhanced T1, T2, and FLAIR imaging sequences, trained and validated with five-fold cross-validation on the 2021 BraTS-RSNA glioma population of 1251 patients, and tested on a diverse, real-world 50 patient sample. Results: Models trained on incomplete data segmented lesions well, often equivalently to those trained on complete data, exhibiting Dice coefficients of 0.907 (single sequence) to 0.945 (full datasets) for whole tumours, and 0.701 (single sequence) to 0.891 (full datasets) for component tissue types. Incomplete data segmentation models could accurately detect enhancing tumour in the absence of contrast imaging, quantifying its volume with an R2 between 0.95-0.97. Conclusions: Deep learning segmentation models characterize tumours well when missing data and can even detect enhancing tissue without the use of contrast. This suggests translation to clinical practice, where incomplete data is common, may be easier than hitherto believed, and may be of value in reducing dependence on contrast use.
Deep forecasting of translational impact in medical research
Oct 17, 2021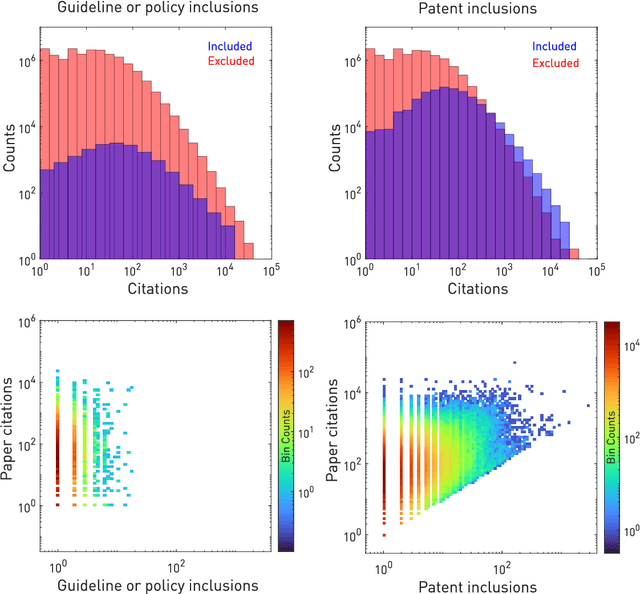
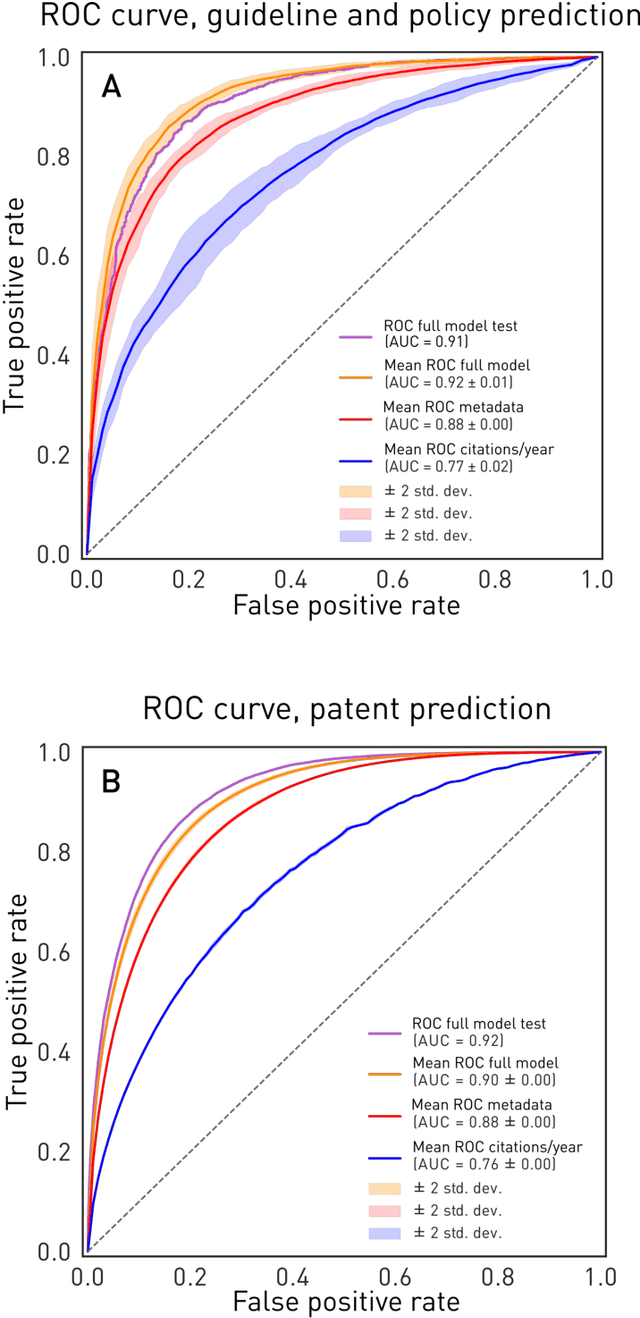
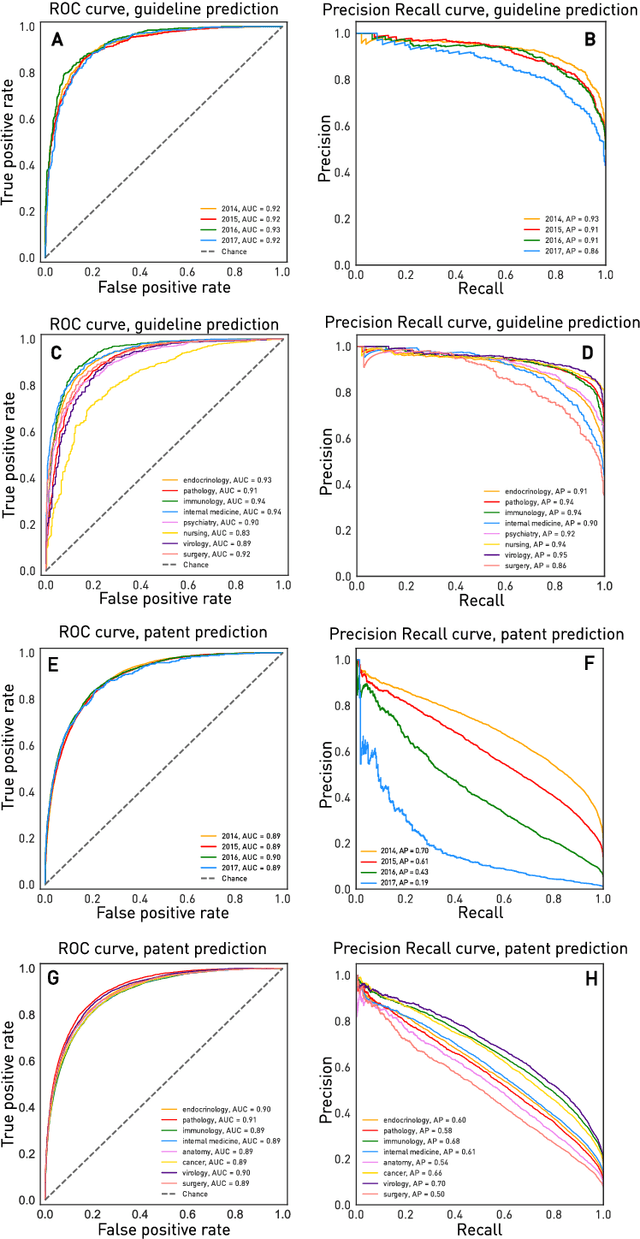
The value of biomedical research--a $1.7 trillion annual investment--is ultimately determined by its downstream, real-world impact. Current objective predictors of impact rest on proxy, reductive metrics of dissemination, such as paper citation rates, whose relation to real-world translation remains unquantified. Here we sought to determine the comparative predictability of future real-world translation--as indexed by inclusion in patents, guidelines or policy documents--from complex models of the abstract-level content of biomedical publications versus citations and publication meta-data alone. We develop a suite of representational and discriminative mathematical models of multi-scale publication data, quantifying predictive performance out-of-sample, ahead-of-time, across major biomedical domains, using the entire corpus of biomedical research captured by Microsoft Academic Graph from 1990 to 2019, encompassing 43.3 million papers across all domains. We show that citations are only moderately predictive of translational impact as judged by inclusion in patents, guidelines, or policy documents. By contrast, high-dimensional models of publication titles, abstracts and metadata exhibit high fidelity (AUROC > 0.9), generalise across time and thematic domain, and transfer to the task of recognising papers of Nobel Laureates. The translational impact of a paper indexed by inclusion in patents, guidelines, or policy documents can be predicted--out-of-sample and ahead-of-time--with substantially higher fidelity from complex models of its abstract-level content than from models of publication meta-data or citation metrics. We argue that content-based models of impact are superior in performance to conventional, citation-based measures, and sustain a stronger evidence-based claim to the objective measurement of translational potential.