 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBidirectional human-AI collaboration in brain tumour assessments improves both expert human and AI agent performance
Dec 13, 2025The benefits of artificial intelligence (AI) human partnerships-evaluating how AI agents enhance expert human performance-are increasingly studied. Though rarely evaluated in healthcare, an inverse approach is possible: AI benefiting from the support of an expert human agent. Here, we investigate both human-AI clinical partnership paradigms in the magnetic resonance imaging-guided characterisation of patients with brain tumours. We reveal that human-AI partnerships improve accuracy and metacognitive ability not only for radiologists supported by AI, but also for AI agents supported by radiologists. Moreover, the greatest patient benefit was evident with an AI agent supported by a human one. Synergistic improvements in agent accuracy, metacognitive performance, and inter-rater agreement suggest that AI can create more capable, confident, and consistent clinical agents, whether human or model-based. Our work suggests that the maximal value of AI in healthcare could emerge not from replacing human intelligence, but from AI agents that routinely leverage and amplify it.
Compressed representation of brain genetic transcription
Oct 24, 2023The architecture of the brain is too complex to be intuitively surveyable without the use of compressed representations that project its variation into a compact, navigable space. The task is especially challenging with high-dimensional data, such as gene expression, where the joint complexity of anatomical and transcriptional patterns demands maximum compression. Established practice is to use standard principal component analysis (PCA), whose computational felicity is offset by limited expressivity, especially at great compression ratios. Employing whole-brain, voxel-wise Allen Brain Atlas transcription data, here we systematically compare compressed representations based on the most widely supported linear and non-linear methods-PCA, kernel PCA, non-negative matrix factorization (NMF), t-stochastic neighbour embedding (t-SNE), uniform manifold approximation and projection (UMAP), and deep auto-encoding-quantifying reconstruction fidelity, anatomical coherence, and predictive utility with respect to signalling, microstructural, and metabolic targets. We show that deep auto-encoders yield superior representations across all metrics of performance and target domains, supporting their use as the reference standard for representing transcription patterns in the human brain.
The legibility of the imaged human brain
Aug 23, 2023

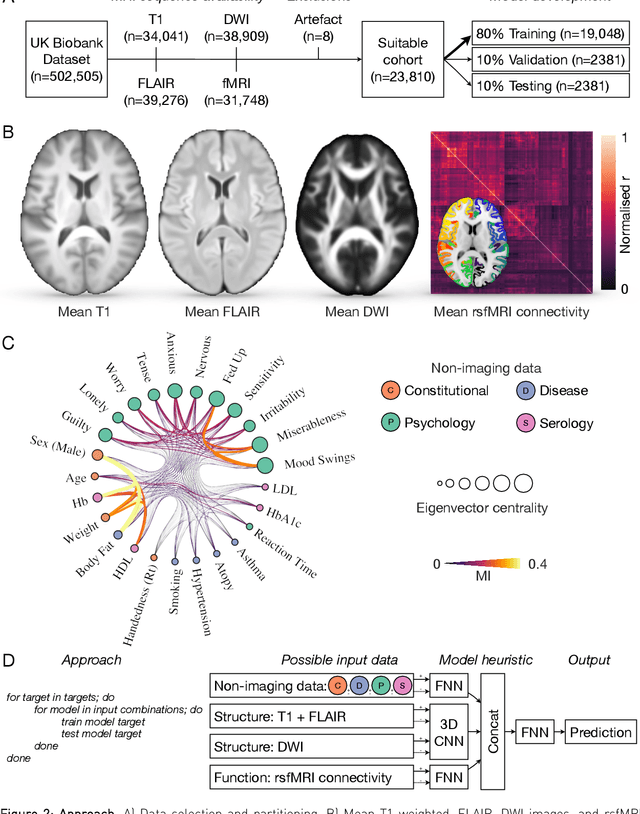

Our knowledge of the organisation of the human brain at the population-level is yet to translate into power to predict functional differences at the individual-level, limiting clinical applications, and casting doubt on the generalisability of inferred mechanisms. It remains unknown whether the difficulty arises from the absence of individuating biological patterns within the brain, or from limited power to access them with the models and compute at our disposal. Here we comprehensively investigate the resolvability of such patterns with data and compute at unprecedented scale. Across 23810 unique participants from UK Biobank, we systematically evaluate the predictability of 25 individual biological characteristics, from all available combinations of structural and functional neuroimaging data. Over 4526 GPU*hours of computation, we train, optimize, and evaluate out-of-sample 700 individual predictive models, including multilayer perceptrons of demographic, psychological, serological, chronic morbidity, and functional connectivity characteristics, and both uni- and multi-modal 3D convolutional neural network models of macro- and micro-structural brain imaging. We find a marked discrepancy between the high predictability of sex (balanced accuracy 99.7%), age (mean absolute error 2.048 years, R2 0.859), and weight (mean absolute error 2.609Kg, R2 0.625), for which we set new state-of-the-art performance, and the surprisingly low predictability of other characteristics. Neither structural nor functional imaging predicted individual psychology better than the coincidence of common chronic morbidity (p<0.05). Serology predicted common morbidity (p<0.05) and was best predicted by it (p<0.001), followed by structural neuroimaging (p<0.05). Our findings suggest either more informative imaging or more powerful models will be needed to decipher individual level characteristics from the brain.
Translating automated brain tumour phenotyping to clinical neuroimaging
Jun 13, 2022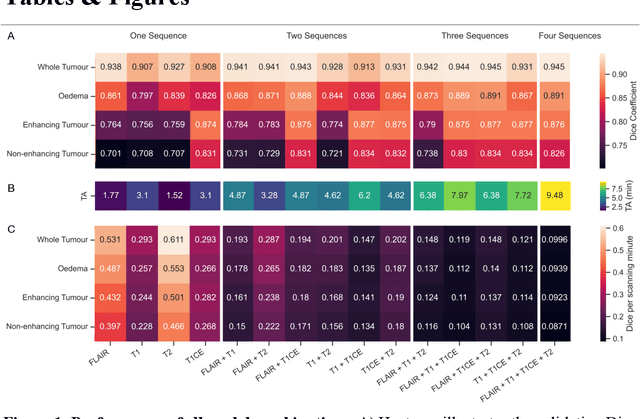
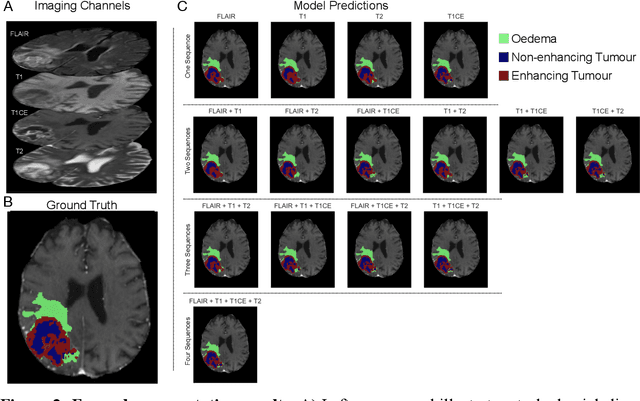


Background: The complex heterogeneity of brain tumours is increasingly recognized to demand data of magnitudes and richness only fully-inclusive, large-scale collections drawn from routine clinical care could plausibly offer. This is a task contemporary machine learning could facilitate, especially in neuroimaging, but its ability to deal with incomplete data common in real world clinical practice remains unknown. Here we apply state-of-the-art methods to large scale, multi-site MRI data to quantify the comparative fidelity of automated tumour segmentation models replicating the various levels of completeness observed in clinical reality. Methods: We compare deep learning (nnU-Net-derived) tumour segmentation models with all possible combinations of T1, contrast-enhanced T1, T2, and FLAIR imaging sequences, trained and validated with five-fold cross-validation on the 2021 BraTS-RSNA glioma population of 1251 patients, and tested on a diverse, real-world 50 patient sample. Results: Models trained on incomplete data segmented lesions well, often equivalently to those trained on complete data, exhibiting Dice coefficients of 0.907 (single sequence) to 0.945 (full datasets) for whole tumours, and 0.701 (single sequence) to 0.891 (full datasets) for component tissue types. Incomplete data segmentation models could accurately detect enhancing tumour in the absence of contrast imaging, quantifying its volume with an R2 between 0.95-0.97. Conclusions: Deep learning segmentation models characterize tumours well when missing data and can even detect enhancing tissue without the use of contrast. This suggests translation to clinical practice, where incomplete data is common, may be easier than hitherto believed, and may be of value in reducing dependence on contrast use.