 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTMA-Grid: An open-source, zero-footprint web application for FAIR Tissue MicroArray De-arraying
Jul 30, 2024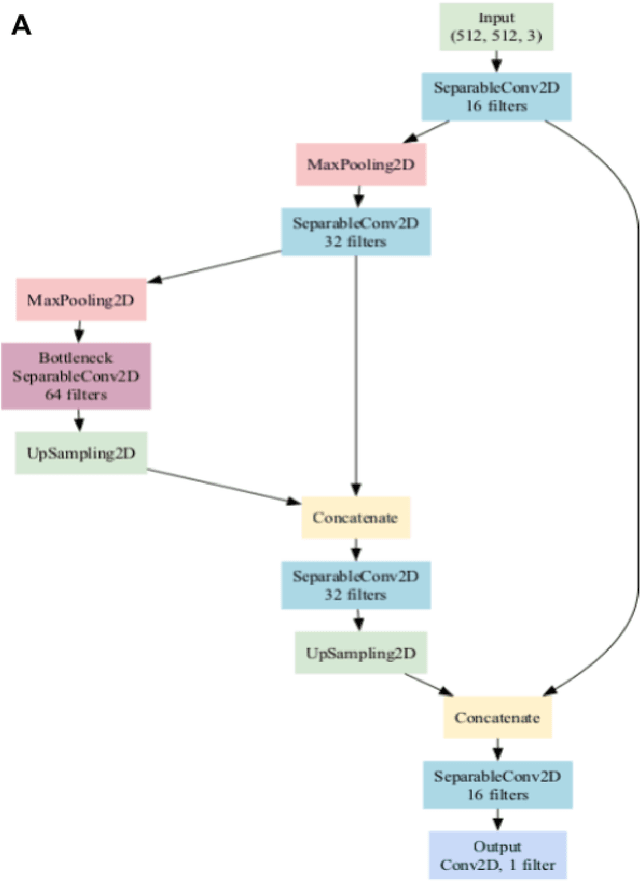


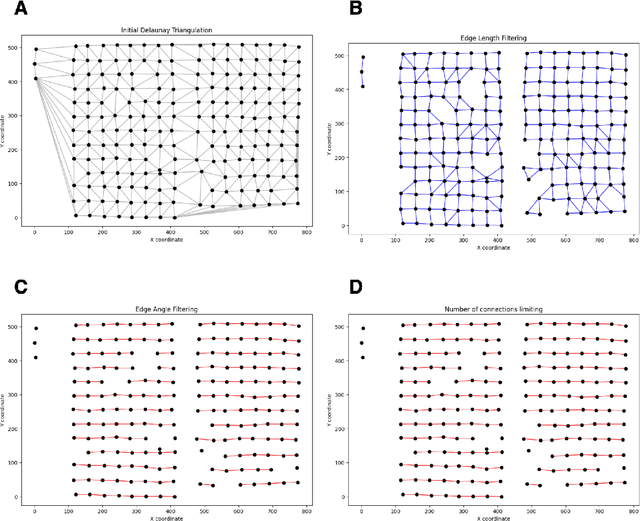
Background: Tissue Microarrays (TMAs) significantly increase analytical efficiency in histopathology and large-scale epidemiologic studies by allowing multiple tissue cores to be scanned on a single slide. The individual cores can be digitally extracted and then linked to metadata for analysis in a process known as de-arraying. However, TMAs often contain core misalignments and artifacts due to assembly errors, which can adversely affect the reliability of the extracted cores during the de-arraying process. Moreover, conventional approaches for TMA de-arraying rely on desktop solutions.Therefore, a robust yet flexible de-arraying method is crucial to account for these inaccuracies and ensure effective downstream analyses. Results: We developed TMA-Grid, an in-browser, zero-footprint, interactive web application for TMA de-arraying. This web application integrates a convolutional neural network for precise tissue segmentation and a grid estimation algorithm to match each identified core to its expected location. The application emphasizes interactivity, allowing users to easily adjust segmentation and gridding results. Operating entirely in the web-browser, TMA-Grid eliminates the need for downloads or installations and ensures data privacy. Adhering to FAIR principles (Findable, Accessible, Interoperable, and Reusable), the application and its components are designed for seamless integration into TMA research workflows. Conclusions: TMA-Grid provides a robust, user-friendly solution for TMA dearraying on the web. As an open, freely accessible platform, it lays the foundation for collaborative analyses of TMAs and similar histopathology imaging data. Availability: Web application: https://episphere.github.io/tma-grid Code: https://github.com/episphere/tma-grid Tutorial: https://youtu.be/miajqyw4BVk
FastImpute: A Baseline for Open-source, Reference-Free Genotype Imputation Methods -- A Case Study in PRS313
Jul 12, 2024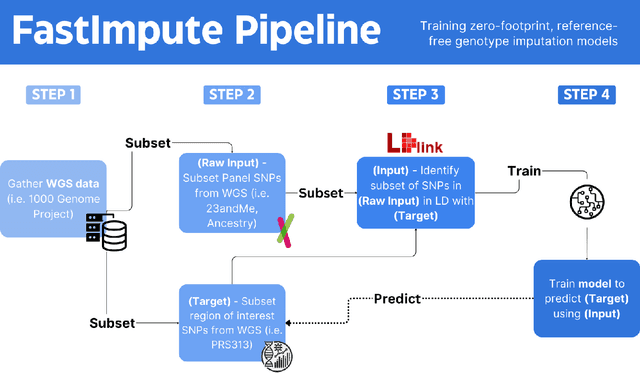
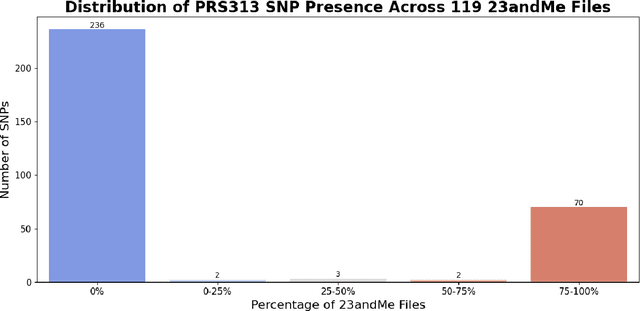
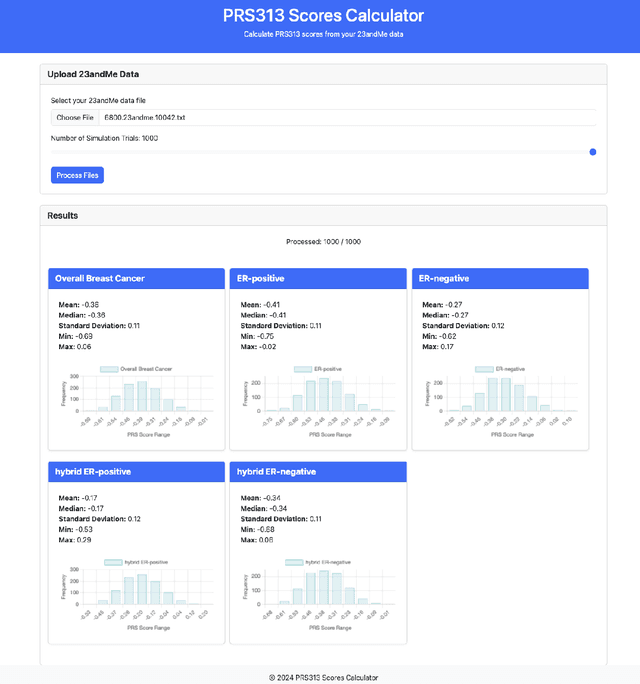
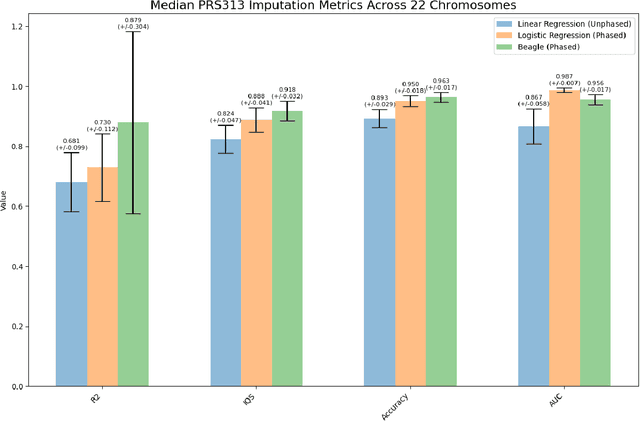
Genotype imputation enhances genetic data by predicting missing SNPs using reference haplotype information. Traditional methods leverage linkage disequilibrium (LD) to infer untyped SNP genotypes, relying on the similarity of LD structures between genotyped target sets and fully sequenced reference panels. Recently, reference-free deep learning-based methods have emerged, offering a promising alternative by predicting missing genotypes without external databases, thereby enhancing privacy and accessibility. However, these methods often produce models with tens of millions of parameters, leading to challenges such as the need for substantial computational resources to train and inefficiency for client-sided deployment. Our study addresses these limitations by introducing a baseline for a novel genotype imputation pipeline that supports client-sided imputation models generalizable across any genotyping chip and genomic region. This approach enhances patient privacy by performing imputation directly on edge devices. As a case study, we focus on PRS313, a polygenic risk score comprising 313 SNPs used for breast cancer risk prediction. Utilizing consumer genetic panels such as 23andMe, our model democratizes access to personalized genetic insights by allowing 23andMe users to obtain their PRS313 score. We demonstrate that simple linear regression can significantly improve the accuracy of PRS313 scores when calculated using SNPs imputed from consumer gene panels, such as 23andMe. Our linear regression model achieved an R^2 of 0.86, compared to 0.33 without imputation and 0.28 with simple imputation (substituting missing SNPs with the minor allele frequency). These findings suggest that popular SNP analysis libraries could benefit from integrating linear regression models for genotype imputation, providing a viable and light-weight alternative to reference based imputation.
Finding Regions of Interest in Whole Slide Images Using Multiple Instance Learning
Apr 11, 2024



Whole Slide Images (WSI), obtained by high-resolution digital scanning of microscope slides at multiple scales, are the cornerstone of modern Digital Pathology. However, they represent a particular challenge to AI-based/AI-mediated analysis because pathology labeling is typically done at slide-level, instead of tile-level. It is not just that medical diagnostics is recorded at the specimen level, the detection of oncogene mutation is also experimentally obtained, and recorded by initiatives like The Cancer Genome Atlas (TCGA), at the slide level. This configures a dual challenge: a) accurately predicting the overall cancer phenotype and b) finding out what cellular morphologies are associated with it at the tile level. To address these challenges, a weakly supervised Multiple Instance Learning (MIL) approach was explored for two prevalent cancer types, Invasive Breast Carcinoma (TCGA-BRCA) and Lung Squamous Cell Carcinoma (TCGA-LUSC). This approach was explored for tumor detection at low magnification levels and TP53 mutations at various levels. Our results show that a novel additive implementation of MIL matched the performance of reference implementation (AUC 0.96), and was only slightly outperformed by Attention MIL (AUC 0.97). More interestingly from the perspective of the molecular pathologist, these different AI architectures identify distinct sensitivities to morphological features (through the detection of Regions of Interest, RoI) at different amplification levels. Tellingly, TP53 mutation was most sensitive to features at the higher applications where cellular morphology is resolved.
Utilizing Automated Breast Cancer Detection to Identify Spatial Distributions of Tumor Infiltrating Lymphocytes in Invasive Breast Cancer
May 29, 2019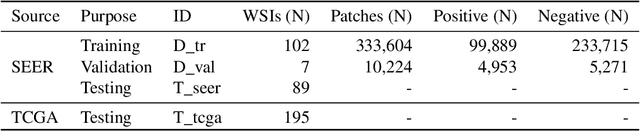
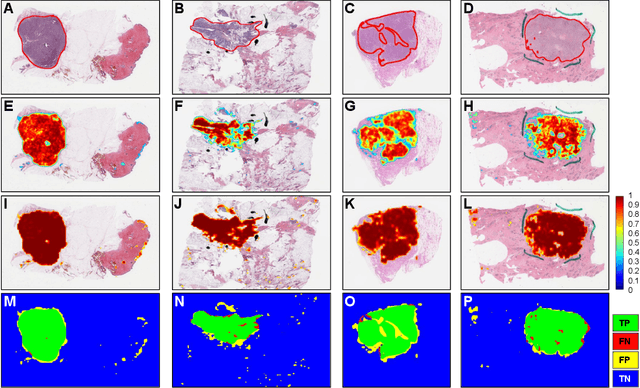
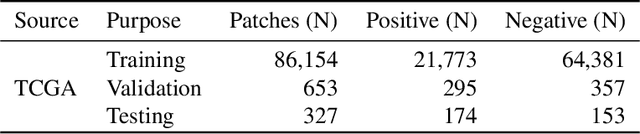
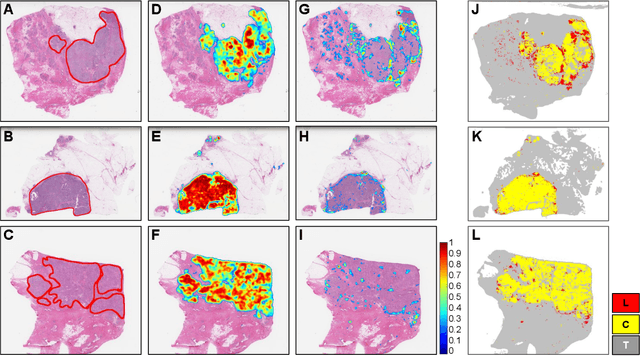
Quantitative assessment of Tumor-TIL spatial relationships is increasingly important in both basic science and clinical aspects of breast cancer research. We have developed and evaluated convolutional neural network (CNN) analysis pipelines to generate combined maps of cancer regions and tumor infiltrating lymphocytes (TILs) in routine diagnostic breast cancer whole slide tissue images (WSIs). We produce interactive whole slide maps that provide 1) insight about the structural patterns and spatial distribution of lymphocytic infiltrates and 2) facilitate improved quantification of TILs. We evaluated both tumor and TIL analyses using three CNN networks - Resnet-34, VGG16 and Inception v4, and demonstrated that the results compared favorably to those obtained by what believe are the best published methods. We have produced open-source tools and generated a public dataset consisting of tumor/TIL maps for 1,015 TCGA breast cancer images. We also present a customized web-based interface that enables easy visualization and interactive exploration of high-resolution combined Tumor-TIL maps for 1,015TCGA invasive breast cancer cases that can be downloaded for further downstream analyses.