 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMemorization in Large Language Models in Medicine: Prevalence, Characteristics, and Implications
Sep 10, 2025Large Language Models (LLMs) have demonstrated significant potential in medicine. To date, LLMs have been widely applied to tasks such as diagnostic assistance, medical question answering, and clinical information synthesis. However, a key open question remains: to what extent do LLMs memorize medical training data. In this study, we present the first comprehensive evaluation of memorization of LLMs in medicine, assessing its prevalence (how frequently it occurs), characteristics (what is memorized), volume (how much content is memorized), and potential downstream impacts (how memorization may affect medical applications). We systematically analyze common adaptation scenarios: (1) continued pretraining on medical corpora, (2) fine-tuning on standard medical benchmarks, and (3) fine-tuning on real-world clinical data, including over 13,000 unique inpatient records from Yale New Haven Health System. The results demonstrate that memorization is prevalent across all adaptation scenarios and significantly higher than reported in the general domain. Memorization affects both the development and adoption of LLMs in medicine and can be categorized into three types: beneficial (e.g., accurate recall of clinical guidelines and biomedical references), uninformative (e.g., repeated disclaimers or templated medical document language), and harmful (e.g., regeneration of dataset-specific or sensitive clinical content). Based on these findings, we offer practical recommendations to facilitate beneficial memorization that enhances domain-specific reasoning and factual accuracy, minimize uninformative memorization to promote deeper learning beyond surface-level patterns, and mitigate harmful memorization to prevent the leakage of sensitive or identifiable patient information.
Generating Synthetic Electronic Health Record (EHR) Data: A Review with Benchmarking
Nov 06, 2024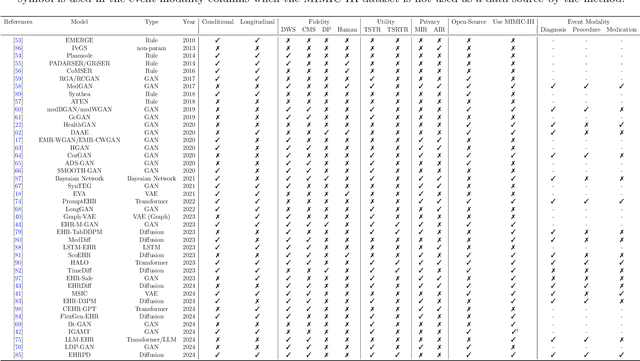
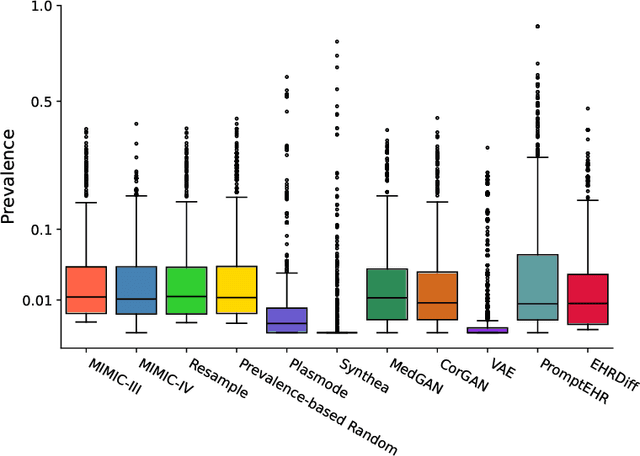
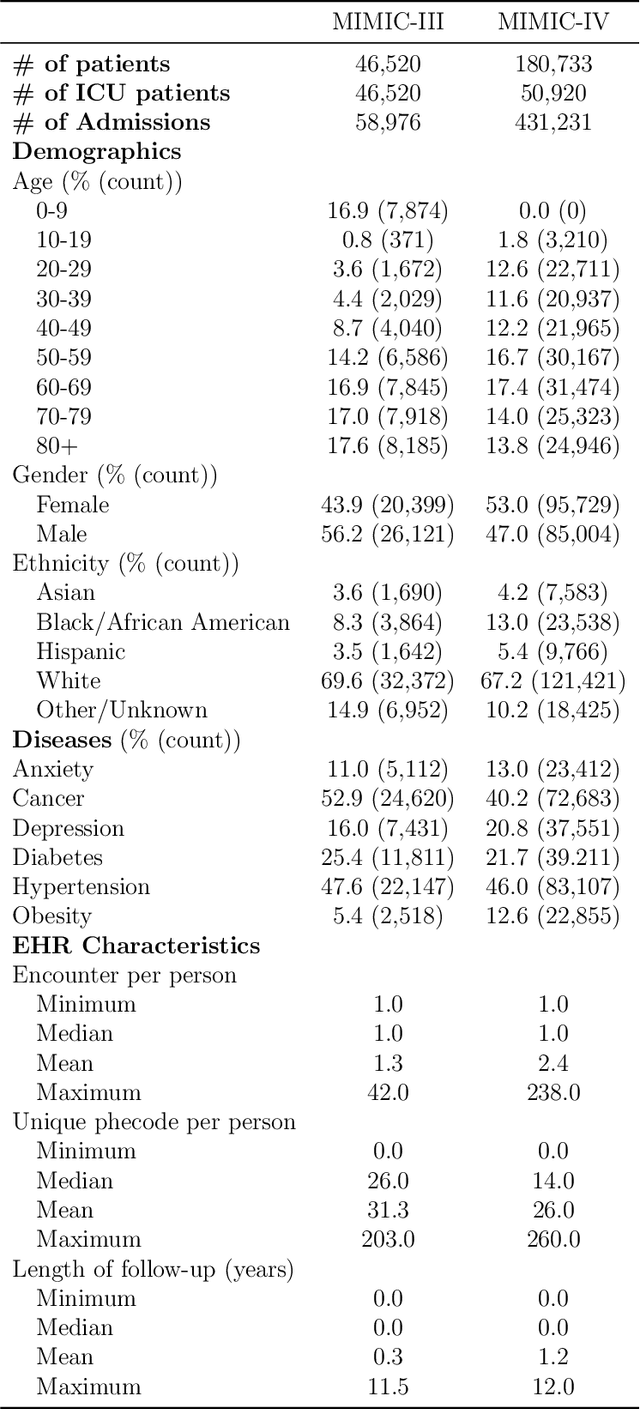
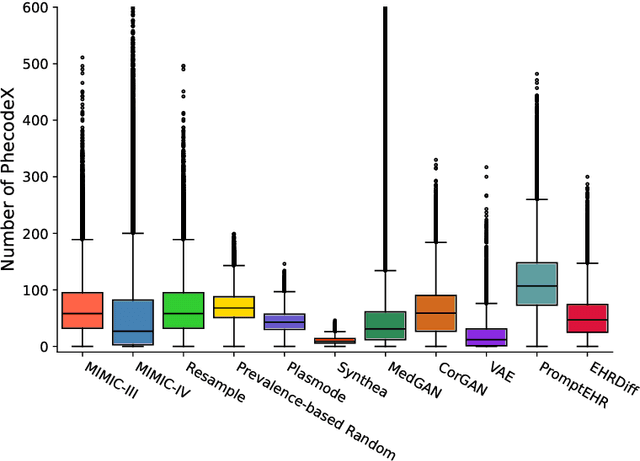
We conduct a scoping review of existing approaches for synthetic EHR data generation, and benchmark major methods with proposed open-source software to offer recommendations for practitioners. We search three academic databases for our scoping review. Methods are benchmarked on open-source EHR datasets, MIMIC-III/IV. Seven existing methods covering major categories and two baseline methods are implemented and compared. Evaluation metrics concern data fidelity, downstream utility, privacy protection, and computational cost. 42 studies are identified and classified into five categories. Seven open-source methods covering all categories are selected, trained on MIMIC-III, and evaluated on MIMIC-III or MIMIC-IV for transportability considerations. Among them, GAN-based methods demonstrate competitive performance in fidelity and utility on MIMIC-III; rule-based methods excel in privacy protection. Similar findings are observed on MIMIC-IV, except that GAN-based methods further outperform the baseline methods in preserving fidelity. A Python package, ``SynthEHRella'', is provided to integrate various choices of approaches and evaluation metrics, enabling more streamlined exploration and evaluation of multiple methods. We found that method choice is governed by the relative importance of the evaluation metrics in downstream use cases. We provide a decision tree to guide the choice among the benchmarked methods. Based on the decision tree, GAN-based methods excel when distributional shifts exist between the training and testing populations. Otherwise, CorGAN and MedGAN are most suitable for association modeling and predictive modeling, respectively. Future research should prioritize enhancing fidelity of the synthetic data while controlling privacy exposure, and comprehensive benchmarking of longitudinal or conditional generation methods.
k-SALSA: k-anonymous synthetic averaging of retinal images via local style alignment
Mar 20, 2023The application of modern machine learning to retinal image analyses offers valuable insights into a broad range of human health conditions beyond ophthalmic diseases. Additionally, data sharing is key to fully realizing the potential of machine learning models by providing a rich and diverse collection of training data. However, the personally-identifying nature of retinal images, encompassing the unique vascular structure of each individual, often prevents this data from being shared openly. While prior works have explored image de-identification strategies based on synthetic averaging of images in other domains (e.g. facial images), existing techniques face difficulty in preserving both privacy and clinical utility in retinal images, as we demonstrate in our work. We therefore introduce k-SALSA, a generative adversarial network (GAN)-based framework for synthesizing retinal fundus images that summarize a given private dataset while satisfying the privacy notion of k-anonymity. k-SALSA brings together state-of-the-art techniques for training and inverting GANs to achieve practical performance on retinal images. Furthermore, k-SALSA leverages a new technique, called local style alignment, to generate a synthetic average that maximizes the retention of fine-grain visual patterns in the source images, thus improving the clinical utility of the generated images. On two benchmark datasets of diabetic retinopathy (EyePACS and APTOS), we demonstrate our improvement upon existing methods with respect to image fidelity, classification performance, and mitigation of membership inference attacks. Our work represents a step toward broader sharing of retinal images for scientific collaboration. Code is available at https://github.com/hcholab/k-salsa.
Mechanisms for Hiding Sensitive Genotypes with Information-Theoretic Privacy
Jul 10, 2020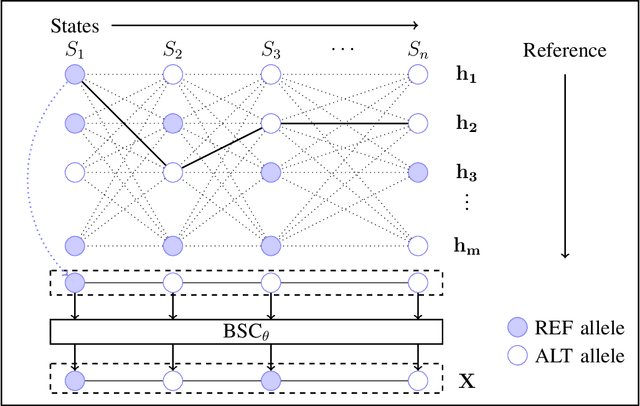
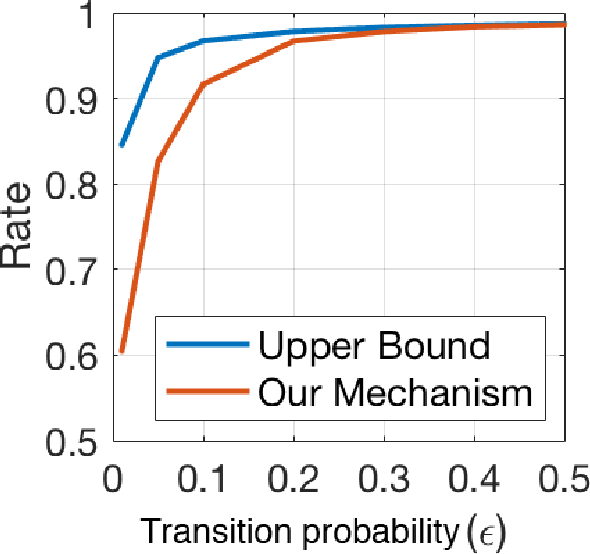
The growing availability of personal genomics services comes with increasing concerns for genomic privacy. Individuals may wish to withhold sensitive genotypes that contain critical health-related information when sharing their data with such services. A straightforward solution that masks only the sensitive genotypes does not ensure privacy due to the correlation structure within the genome. Here, we develop an information-theoretic mechanism for masking sensitive genotypes, which ensures no information about the sensitive genotypes is leaked. We also propose an efficient algorithmic implementation of our mechanism for genomic data governed by hidden Markov models. Our work is a step towards more rigorous control of privacy in genomic data sharing.
Large-Margin Classification in Hyperbolic Space
Jun 01, 2018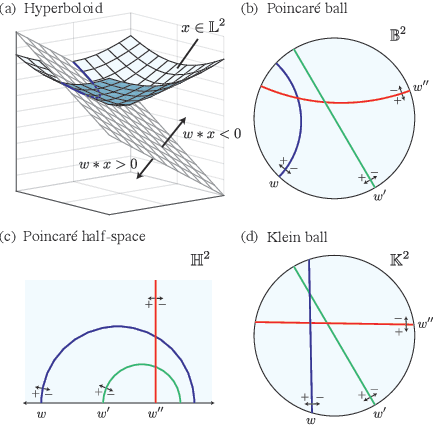
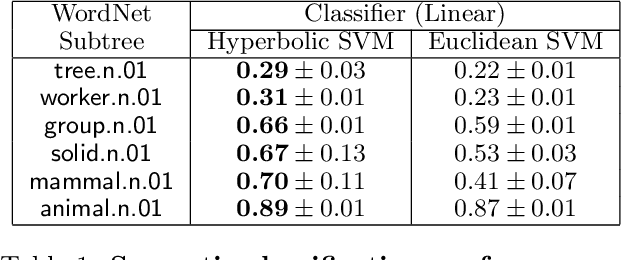
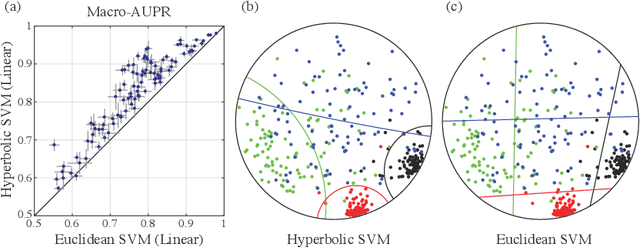
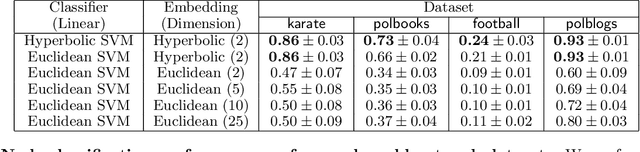
Representing data in hyperbolic space can effectively capture latent hierarchical relationships. With the goal of enabling accurate classification of points in hyperbolic space while respecting their hyperbolic geometry, we introduce hyperbolic SVM, a hyperbolic formulation of support vector machine classifiers, and elucidate through new theoretical work its connection to the Euclidean counterpart. We demonstrate the performance improvement of hyperbolic SVM for multi-class prediction tasks on real-world complex networks as well as simulated datasets. Our work allows analytic pipelines that take the inherent hyperbolic geometry of the data into account in an end-to-end fashion without resorting to ill-fitting tools developed for Euclidean space.
Diffusion Component Analysis: Unraveling Functional Topology in Biological Networks
Apr 10, 2015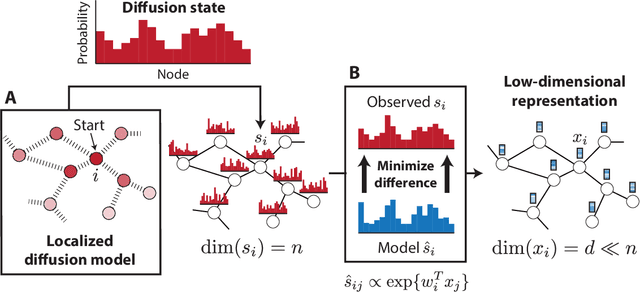
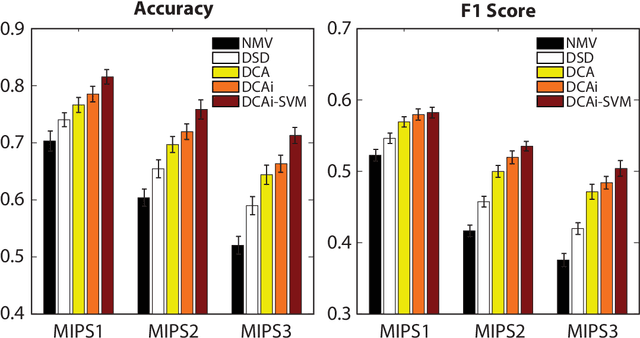
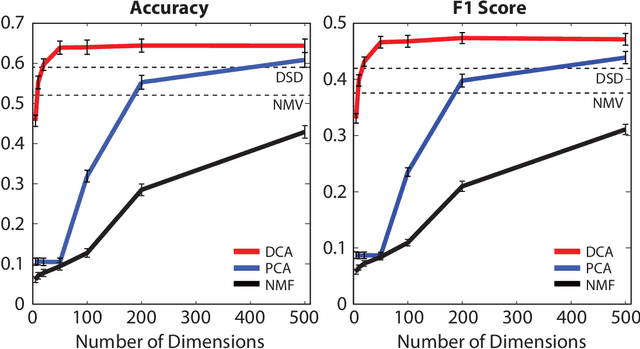
Complex biological systems have been successfully modeled by biochemical and genetic interaction networks, typically gathered from high-throughput (HTP) data. These networks can be used to infer functional relationships between genes or proteins. Using the intuition that the topological role of a gene in a network relates to its biological function, local or diffusion based "guilt-by-association" and graph-theoretic methods have had success in inferring gene functions. Here we seek to improve function prediction by integrating diffusion-based methods with a novel dimensionality reduction technique to overcome the incomplete and noisy nature of network data. In this paper, we introduce diffusion component analysis (DCA), a framework that plugs in a diffusion model and learns a low-dimensional vector representation of each node to encode the topological properties of a network. As a proof of concept, we demonstrate DCA's substantial improvement over state-of-the-art diffusion-based approaches in predicting protein function from molecular interaction networks. Moreover, our DCA framework can integrate multiple networks from heterogeneous sources, consisting of genomic information, biochemical experiments and other resources, to even further improve function prediction. Yet another layer of performance gain is achieved by integrating the DCA framework with support vector machines that take our node vector representations as features. Overall, our DCA framework provides a novel representation of nodes in a network that can be used as a plug-in architecture to other machine learning algorithms to decipher topological properties of and obtain novel insights into interactomes.