 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Representation Learning on Tissue-Specific Multi-Omics
Jul 25, 2021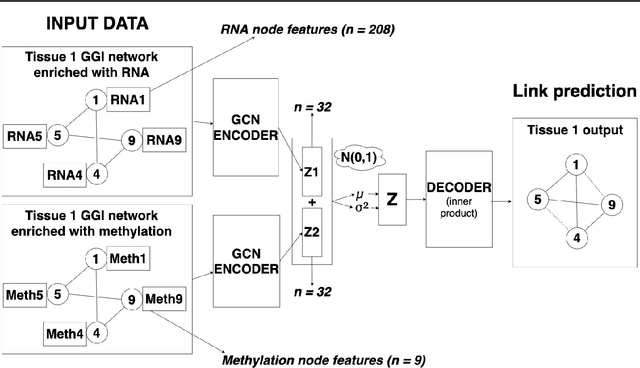
Combining different modalities of data from human tissues has been critical in advancing biomedical research and personalised medical care. In this study, we leverage a graph embedding model (i.e VGAE) to perform link prediction on tissue-specific Gene-Gene Interaction (GGI) networks. Through ablation experiments, we prove that the combination of multiple biological modalities (i.e multi-omics) leads to powerful embeddings and better link prediction performances. Our evaluation shows that the integration of gene methylation profiles and RNA-sequencing data significantly improves the link prediction performance. Overall, the combination of RNA-sequencing and gene methylation data leads to a link prediction accuracy of 71% on GGI networks. By harnessing graph representation learning on multi-omics data, our work brings novel insights to the current literature on multi-omics integration in bioinformatics.
Is Disentanglement all you need? Comparing Concept-based & Disentanglement Approaches
Apr 14, 2021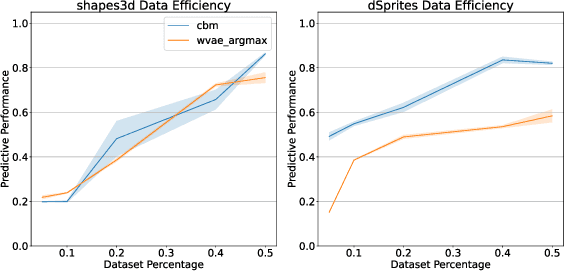

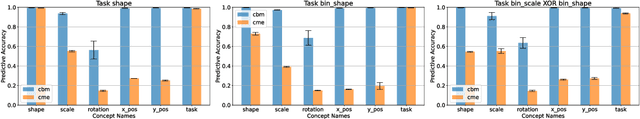

Concept-based explanations have emerged as a popular way of extracting human-interpretable representations from deep discriminative models. At the same time, the disentanglement learning literature has focused on extracting similar representations in an unsupervised or weakly-supervised way, using deep generative models. Despite the overlapping goals and potential synergies, to our knowledge, there has not yet been a systematic comparison of the limitations and trade-offs between concept-based explanations and disentanglement approaches. In this paper, we give an overview of these fields, comparing and contrasting their properties and behaviours on a diverse set of tasks, and highlighting their potential strengths and limitations. In particular, we demonstrate that state-of-the-art approaches from both classes can be data inefficient, sensitive to the specific nature of the classification/regression task, or sensitive to the employed concept representation.
Using ontology embeddings for structural inductive bias in gene expression data analysis
Nov 22, 2020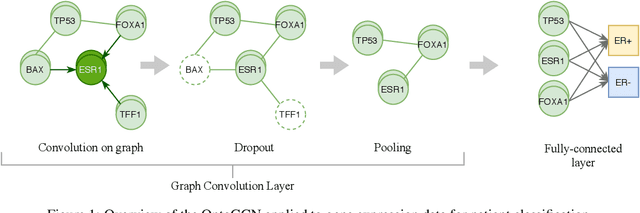

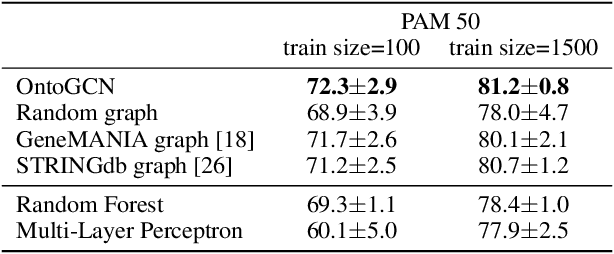
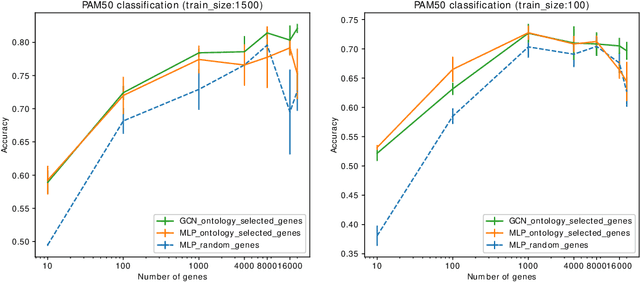
Stratifying cancer patients based on their gene expression levels allows improving diagnosis, survival analysis and treatment planning. However, such data is extremely highly dimensional as it contains expression values for over 20000 genes per patient, and the number of samples in the datasets is low. To deal with such settings, we propose to incorporate prior biological knowledge about genes from ontologies into the machine learning system for the task of patient classification given their gene expression data. We use ontology embeddings that capture the semantic similarities between the genes to direct a Graph Convolutional Network, and therefore sparsify the network connections. We show this approach provides an advantage for predicting clinical targets from high-dimensional low-sample data.
Incorporating network based protein complex discovery into automated model construction
Sep 29, 2020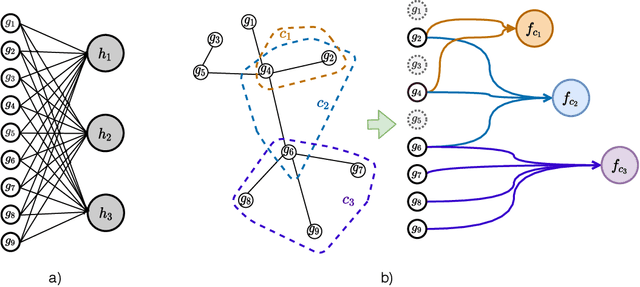
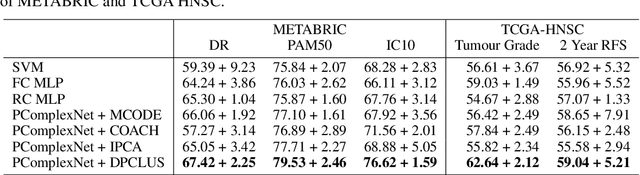
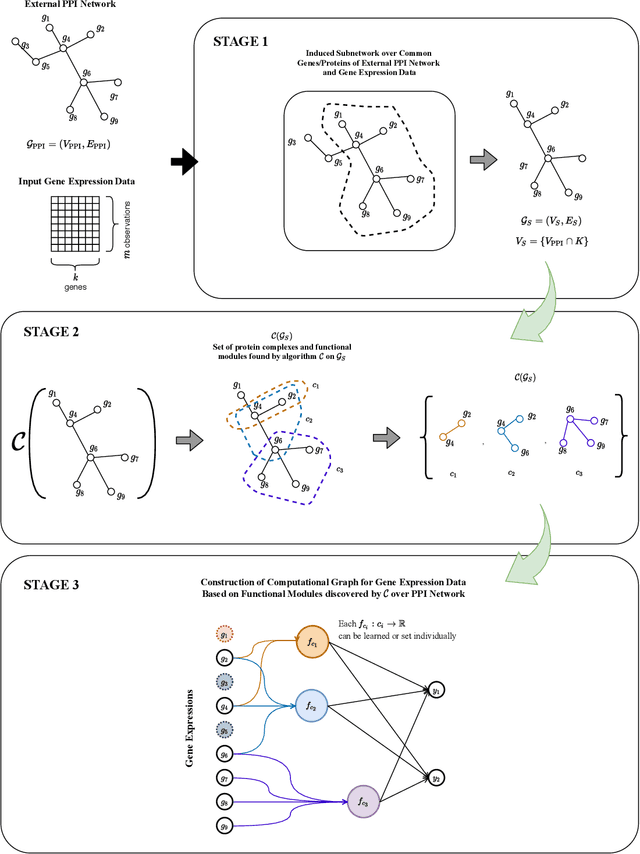
We propose a method for gene expression based analysis of cancer phenotypes incorporating network biology knowledge through unsupervised construction of computational graphs. The structural construction of the computational graphs is driven by the use of topological clustering algorithms on protein-protein networks which incorporate inductive biases stemming from network biology research in protein complex discovery. This structurally constrains the hypothesis space over the possible computational graph factorisation whose parameters can then be learned through supervised or unsupervised task settings. The sparse construction of the computational graph enables the differential protein complex activity analysis whilst also interpreting the individual contributions of genes/proteins involved in each individual protein complex. In our experiments analysing a variety of cancer phenotypes, we show that the proposed methods outperform SVM, Fully-Connected MLP, and Randomly-Connected MLPs in all tasks. Our work introduces a scalable method for incorporating large interaction networks as prior knowledge to drive the construction of powerful computational models amenable to introspective study.