 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIncorporating network based protein complex discovery into automated model construction
Sep 29, 2020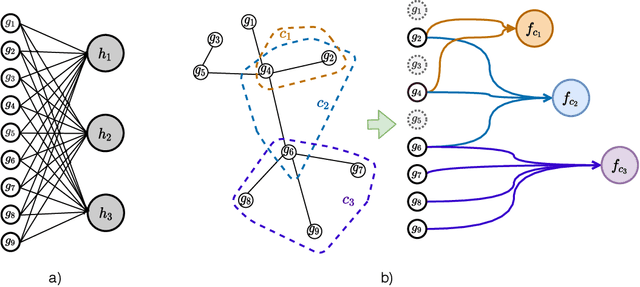
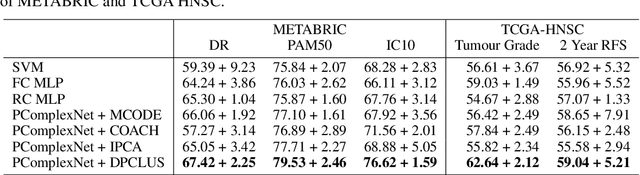
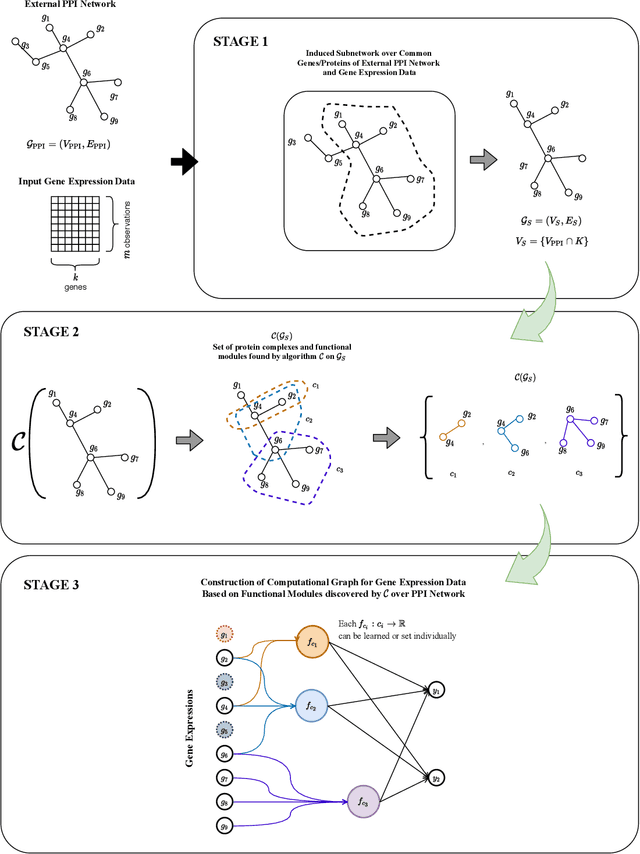
We propose a method for gene expression based analysis of cancer phenotypes incorporating network biology knowledge through unsupervised construction of computational graphs. The structural construction of the computational graphs is driven by the use of topological clustering algorithms on protein-protein networks which incorporate inductive biases stemming from network biology research in protein complex discovery. This structurally constrains the hypothesis space over the possible computational graph factorisation whose parameters can then be learned through supervised or unsupervised task settings. The sparse construction of the computational graph enables the differential protein complex activity analysis whilst also interpreting the individual contributions of genes/proteins involved in each individual protein complex. In our experiments analysing a variety of cancer phenotypes, we show that the proposed methods outperform SVM, Fully-Connected MLP, and Randomly-Connected MLPs in all tasks. Our work introduces a scalable method for incorporating large interaction networks as prior knowledge to drive the construction of powerful computational models amenable to introspective study.