 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeToward Guarantees for Clinical Reasoning in Vision Language Models via Formal Verification
Feb 27, 2026Vision-language models (VLMs) show promise in drafting radiology reports, yet they frequently suffer from logical inconsistencies, generating diagnostic impressions unsupported by their own perceptual findings or missing logically entailed conclusions. Standard lexical metrics heavily penalize clinical paraphrasing and fail to capture these deductive failures in reference-free settings. Toward guarantees for clinical reasoning, we introduce a neurosymbolic verification framework that deterministically audits the internal consistency of VLM-generated reports. Our pipeline autoformalizes free-text radiographic findings into structured propositional evidence, utilizing an SMT solver (Z3) and a clinical knowledge base to verify whether each diagnostic claim is mathematically entailed, hallucinated, or omitted. Evaluating seven VLMs across five chest X-ray benchmarks, our verifier exposes distinct reasoning failure modes, such as conservative observation and stochastic hallucination, that remain invisible to traditional metrics. On labeled datasets, enforcing solver-backed entailment acts as a rigorous post-hoc guarantee, systematically eliminating unsupported hallucinations to significantly increase diagnostic soundness and precision in generative clinical assistants.
Trust The Typical
Feb 04, 2026Current approaches to LLM safety fundamentally rely on a brittle cat-and-mouse game of identifying and blocking known threats via guardrails. We argue for a fresh approach: robust safety comes not from enumerating what is harmful, but from deeply understanding what is safe. We introduce Trust The Typical (T3), a framework that operationalizes this principle by treating safety as an out-of-distribution (OOD) detection problem. T3 learns the distribution of acceptable prompts in a semantic space and flags any significant deviation as a potential threat. Unlike prior methods, it requires no training on harmful examples, yet achieves state-of-the-art performance across 18 benchmarks spanning toxicity, hate speech, jailbreaking, multilingual harms, and over-refusal, reducing false positive rates by up to 40x relative to specialized safety models. A single model trained only on safe English text transfers effectively to diverse domains and over 14 languages without retraining. Finally, we demonstrate production readiness by integrating a GPU-optimized version into vLLM, enabling continuous guardrailing during token generation with less than 6% overhead even under dense evaluation intervals on large-scale workloads.
HugRAG: Hierarchical Causal Knowledge Graph Design for RAG
Feb 04, 2026Retrieval augmented generation (RAG) has enhanced large language models by enabling access to external knowledge, with graph-based RAG emerging as a powerful paradigm for structured retrieval and reasoning. However, existing graph-based methods often over-rely on surface-level node matching and lack explicit causal modeling, leading to unfaithful or spurious answers. Prior attempts to incorporate causality are typically limited to local or single-document contexts and also suffer from information isolation that arises from modular graph structures, which hinders scalability and cross-module causal reasoning. To address these challenges, we propose HugRAG, a framework that rethinks knowledge organization for graph-based RAG through causal gating across hierarchical modules. HugRAG explicitly models causal relationships to suppress spurious correlations while enabling scalable reasoning over large-scale knowledge graphs. Extensive experiments demonstrate that HugRAG consistently outperforms competitive graph-based RAG baselines across multiple datasets and evaluation metrics. Our work establishes a principled foundation for structured, scalable, and causally grounded RAG systems.
VERGE: Formal Refinement and Guidance Engine for Verifiable LLM Reasoning
Jan 27, 2026Despite the syntactic fluency of Large Language Models (LLMs), ensuring their logical correctness in high-stakes domains remains a fundamental challenge. We present a neurosymbolic framework that combines LLMs with SMT solvers to produce verification-guided answers through iterative refinement. Our approach decomposes LLM outputs into atomic claims, autoformalizes them into first-order logic, and verifies their logical consistency using automated theorem proving. We introduce three key innovations: (1) multi-model consensus via formal semantic equivalence checking to ensure logic-level alignment between candidates, eliminating the syntactic bias of surface-form metrics, (2) semantic routing that directs different claim types to appropriate verification strategies: symbolic solvers for logical claims and LLM ensembles for commonsense reasoning, and (3) precise logical error localization via Minimal Correction Subsets (MCS), which pinpoint the exact subset of claims to revise, transforming binary failure signals into actionable feedback. Our framework classifies claims by their logical status and aggregates multiple verification signals into a unified score with variance-based penalty. The system iteratively refines answers using structured feedback until acceptance criteria are met or convergence is achieved. This hybrid approach delivers formal guarantees where possible and consensus verification elsewhere, advancing trustworthy AI. With the GPT-OSS-120B model, VERGE demonstrates an average performance uplift of 18.7% at convergence across a set of reasoning benchmarks compared to single-pass approaches.
Mid-Think: Training-Free Intermediate-Budget Reasoning via Token-Level Triggers
Jan 11, 2026Hybrid reasoning language models are commonly controlled through high-level Think/No-think instructions to regulate reasoning behavior, yet we found that such mode switching is largely driven by a small set of trigger tokens rather than the instructions themselves. Through attention analysis and controlled prompting experiments, we show that a leading ``Okay'' token induces reasoning behavior, while the newline pattern following ``</think>'' suppresses it. Based on this observation, we propose Mid-Think, a simple training-free prompting format that combines these triggers to achieve intermediate-budget reasoning, consistently outperforming fixed-token and prompt-based baselines in terms of the accuracy-length trade-off. Furthermore, applying Mid-Think to RL training after SFT reduces training time by approximately 15% while improving final performance of Qwen3-8B on AIME from 69.8% to 72.4% and on GPQA from 58.5% to 61.1%, demonstrating its effectiveness for both inference-time control and RL-based reasoning training.
$K^4$: Online Log Anomaly Detection Via Unsupervised Typicality Learning
Jul 26, 2025



Existing Log Anomaly Detection (LogAD) methods are often slow, dependent on error-prone parsing, and use unrealistic evaluation protocols. We introduce $K^4$, an unsupervised and parser-independent framework for high-performance online detection. $K^4$ transforms arbitrary log embeddings into compact four-dimensional descriptors (Precision, Recall, Density, Coverage) using efficient k-nearest neighbor (k-NN) statistics. These descriptors enable lightweight detectors to accurately score anomalies without retraining. Using a more realistic online evaluation protocol, $K^4$ sets a new state-of-the-art (AUROC: 0.995-0.999), outperforming baselines by large margins while being orders of magnitude faster, with training under 4 seconds and inference as low as 4 $\mu$s.
Grammars of Formal Uncertainty: When to Trust LLMs in Automated Reasoning Tasks
May 26, 2025Large language models (LLMs) show remarkable promise for democratizing automated reasoning by generating formal specifications. However, a fundamental tension exists: LLMs are probabilistic, while formal verification demands deterministic guarantees. This paper addresses this epistemological gap by comprehensively investigating failure modes and uncertainty quantification (UQ) in LLM-generated formal artifacts. Our systematic evaluation of five frontier LLMs reveals Satisfiability Modulo Theories (SMT) based autoformalization's domain-specific impact on accuracy (from +34.8% on logical tasks to -44.5% on factual ones), with known UQ techniques like the entropy of token probabilities failing to identify these errors. We introduce a probabilistic context-free grammar (PCFG) framework to model LLM outputs, yielding a refined uncertainty taxonomy. We find uncertainty signals are task-dependent (e.g., grammar entropy for logic, AUROC>0.93). Finally, a lightweight fusion of these signals enables selective verification, drastically reducing errors (14-100%) with minimal abstention, transforming LLM-driven formalization into a reliable engineering discipline.
Bayesian Binary Search
Oct 02, 2024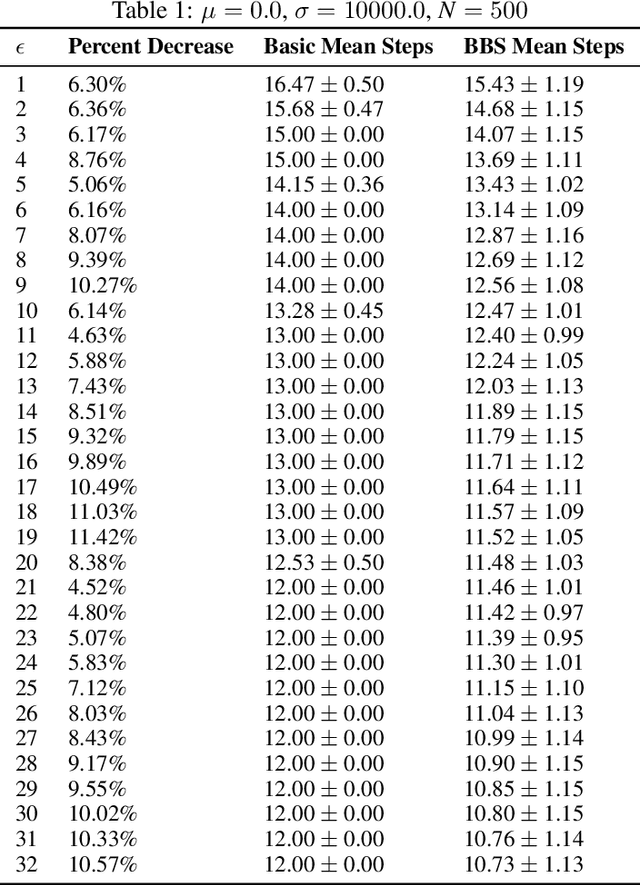
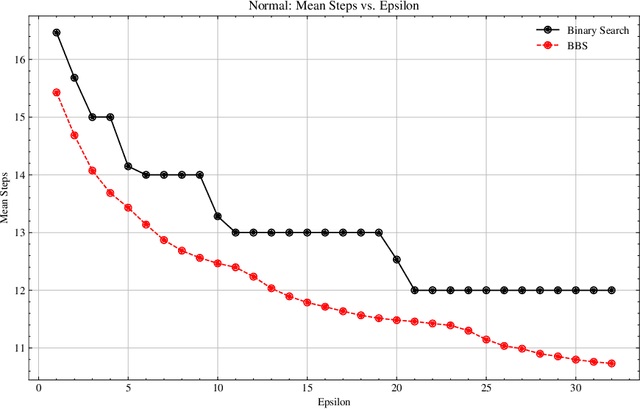

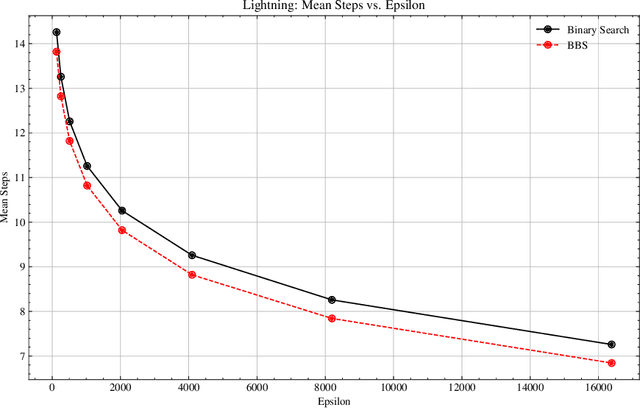
We present Bayesian Binary Search (BBS), a novel probabilistic variant of the classical binary search/bisection algorithm. BBS leverages machine learning/statistical techniques to estimate the probability density of the search space and modifies the bisection step to split based on probability density rather than the traditional midpoint, allowing for the learned distribution of the search space to guide the search algorithm. Search space density estimation can flexibly be performed using supervised probabilistic machine learning techniques (e.g., Gaussian process regression, Bayesian neural networks, quantile regression) or unsupervised learning algorithms (e.g., Gaussian mixture models, kernel density estimation (KDE), maximum likelihood estimation (MLE)). We demonstrate significant efficiency gains of using BBS on both simulated data across a variety of distributions and in a real-world binary search use case of probing channel balances in the Bitcoin Lightning Network, for which we have deployed the BBS algorithm in a production setting.
Channel Balance Interpolation in the Lightning Network via Machine Learning
May 20, 2024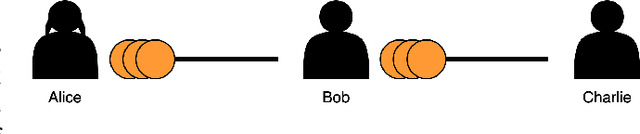

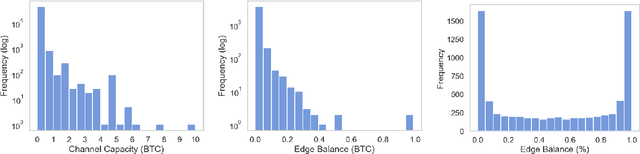
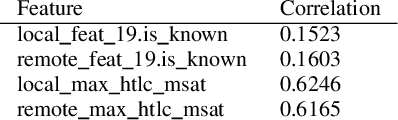
The Bitcoin Lightning Network is a Layer 2 payment protocol that addresses Bitcoin's scalability by facilitating quick and cost effective transactions through payment channels. This research explores the feasibility of using machine learning models to interpolate channel balances within the network, which can be used for optimizing the network's pathfinding algorithms. While there has been much exploration in balance probing and multipath payment protocols, predicting channel balances using solely node and channel features remains an uncharted area. This paper evaluates the performance of several machine learning models against two heuristic baselines and investigates the predictive capabilities of various features. Our model performs favorably in experimental evaluation, outperforming by 10% against an equal split baseline where both edges are assigned half of the channel capacity.
GCExplainer: Human-in-the-Loop Concept-based Explanations for Graph Neural Networks
Jul 25, 2021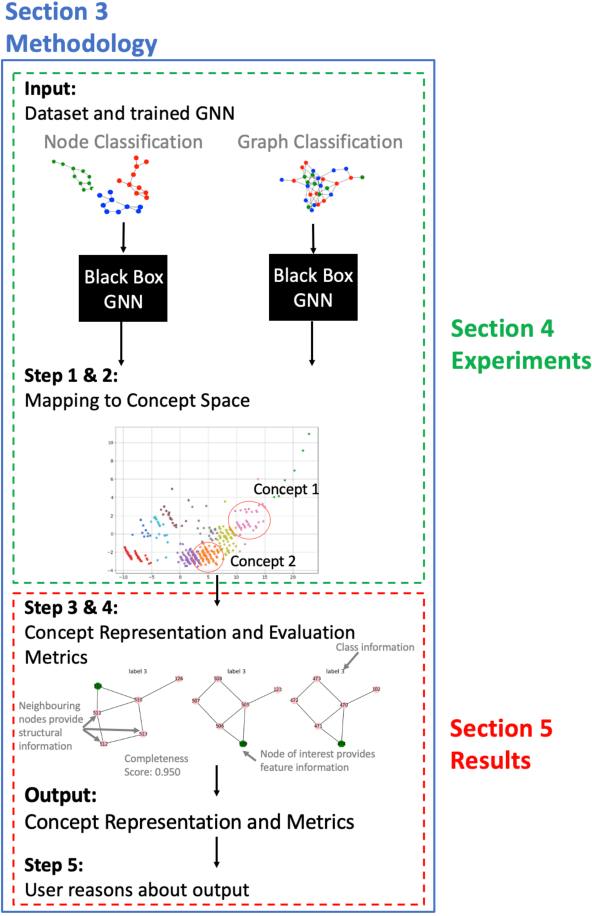
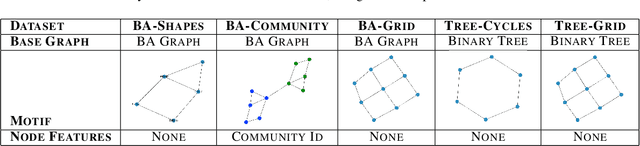
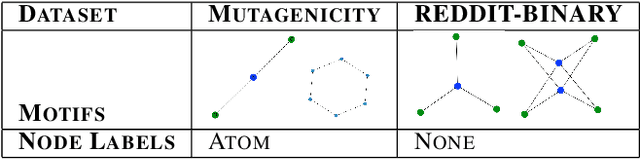
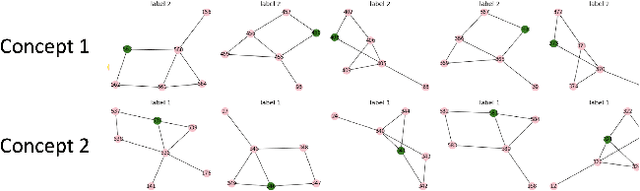
While graph neural networks (GNNs) have been shown to perform well on graph-based data from a variety of fields, they suffer from a lack of transparency and accountability, which hinders trust and consequently the deployment of such models in high-stake and safety-critical scenarios. Even though recent research has investigated methods for explaining GNNs, these methods are limited to single-instance explanations, also known as local explanations. Motivated by the aim of providing global explanations, we adapt the well-known Automated Concept-based Explanation approach (Ghorbani et al., 2019) to GNN node and graph classification, and propose GCExplainer. GCExplainer is an unsupervised approach for post-hoc discovery and extraction of global concept-based explanations for GNNs, which puts the human in the loop. We demonstrate the success of our technique on five node classification datasets and two graph classification datasets, showing that we are able to discover and extract high-quality concept representations by putting the human in the loop. We achieve a maximum completeness score of 1 and an average completeness score of 0.753 across the datasets. Finally, we show that the concept-based explanations provide an improved insight into the datasets and GNN models compared to the state-of-the-art explanations produced by GNNExplainer (Ying et al., 2019).