 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom noisy observables to accurate ground state energies: a quantum classical signal subspace approach with denoising
May 15, 2025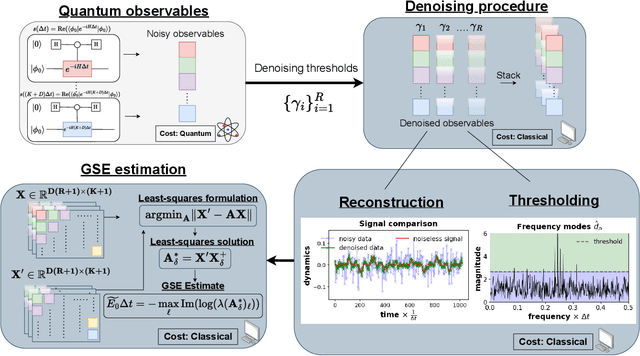
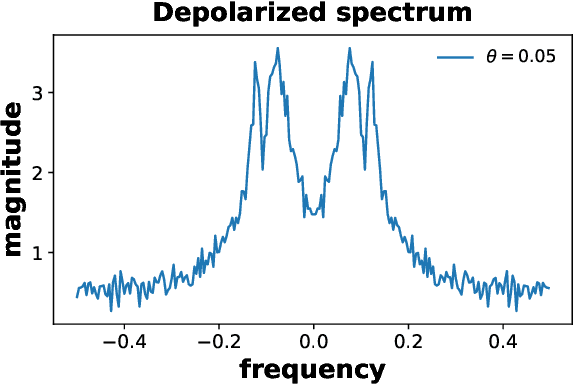
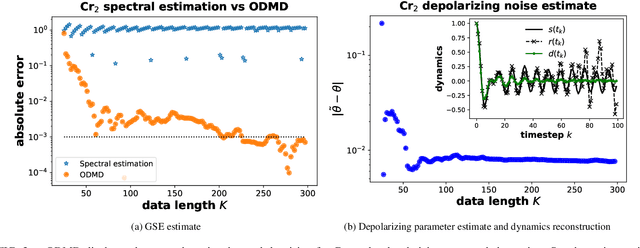
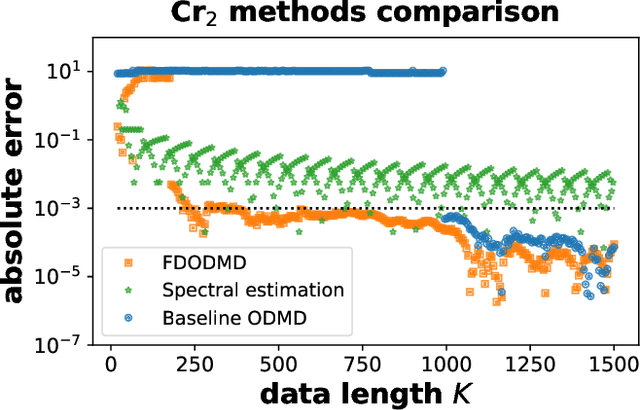
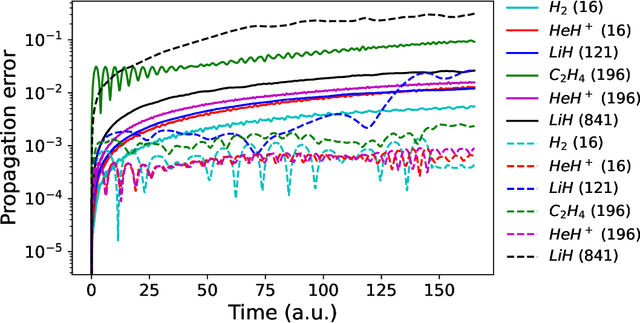
We propose a hybrid quantum-classical algorithm for ground state energy (GSE) estimation that remains robust to highly noisy data and exhibits low sensitivity to hyperparameter tuning. Our approach -- Fourier Denoising Observable Dynamic Mode Decomposition (FDODMD) -- combines Fourier-based denoising thresholding to suppress spurious noise modes with observable dynamic mode decomposition (ODMD), a quantum-classical signal subspace method. By applying ODMD to an ensemble of denoised time-domain trajectories, FDODMD reliably estimates the system's eigenfrequencies. We also provide an error analysis of FDODMD. Numerical experiments on molecular systems demonstrate that FDODMD achieves convergence in high-noise regimes inaccessible to baseline methods under a limited quantum computational budget, while accelerating spectral estimation in intermediate-noise regimes. Importantly, this performance gain is entirely classical, requiring no additional quantum overhead and significantly reducing overall quantum resource demands.
Nonlinear Optimal Control of Electron Dynamics within Hartree-Fock Theory
Dec 04, 2024Consider the problem of determining the optimal applied electric field to drive a molecule from an initial state to a desired target state. For even moderately sized molecules, solving this problem directly using the exact equations of motion -- the time-dependent Schr\"odinger equation (TDSE) -- is numerically intractable. We present a solution of this problem within time-dependent Hartree-Fock (TDHF) theory, a mean field approximation of the TDSE. Optimality is defined in terms of minimizing the total control effort while maximizing the overlap between desired and achieved target states. We frame this problem as an optimization problem constrained by the nonlinear TDHF equations; we solve it using trust region optimization with gradients computed via a custom-built adjoint state method. For three molecular systems, we show that with very small neural network parametrizations of the control, our method yields solutions that achieve desired targets within acceptable constraints and tolerances.
Scalable learning of potentials to predict time-dependent Hartree-Fock dynamics
Aug 08, 2024We propose a framework to learn the time-dependent Hartree-Fock (TDHF) inter-electronic potential of a molecule from its electron density dynamics. Though the entire TDHF Hamiltonian, including the inter-electronic potential, can be computed from first principles, we use this problem as a testbed to develop strategies that can be applied to learn \emph{a priori} unknown terms that arise in other methods/approaches to quantum dynamics, e.g., emerging problems such as learning exchange-correlation potentials for time-dependent density functional theory. We develop, train, and test three models of the TDHF inter-electronic potential, each parameterized by a four-index tensor of size up to $60 \times 60 \times 60 \times 60$. Two of the models preserve Hermitian symmetry, while one model preserves an eight-fold permutation symmetry that implies Hermitian symmetry. Across seven different molecular systems, we find that accounting for the deeper eight-fold symmetry leads to the best-performing model across three metrics: training efficiency, test set predictive power, and direct comparison of true and learned inter-electronic potentials. All three models, when trained on ensembles of field-free trajectories, generate accurate electron dynamics predictions even in a field-on regime that lies outside the training set. To enable our models to scale to large molecular systems, we derive expressions for Jacobian-vector products that enable iterative, matrix-free training.
Neural Continuous-Time Markov Models
Dec 11, 2022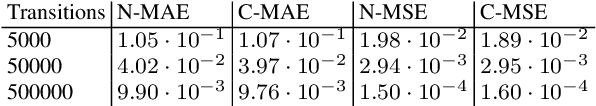
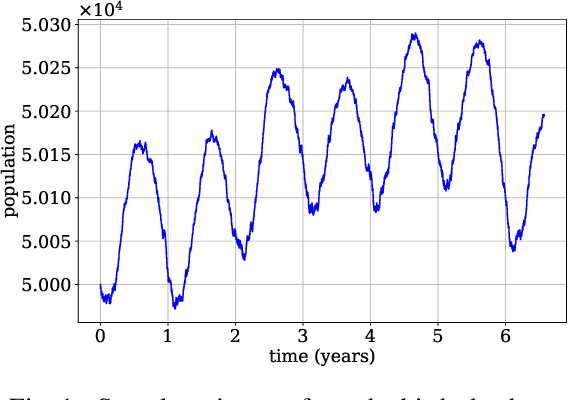
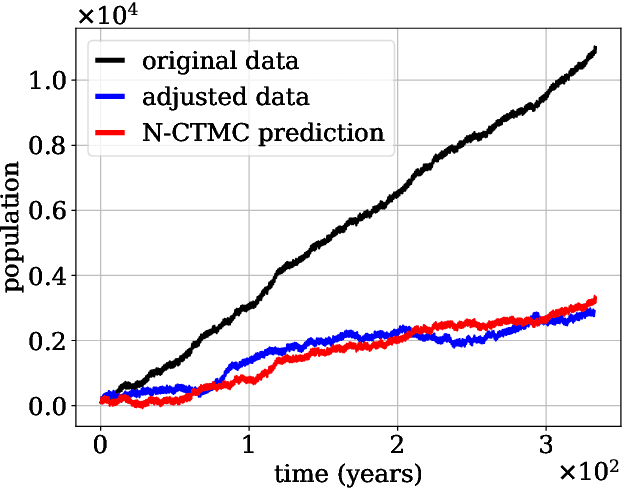
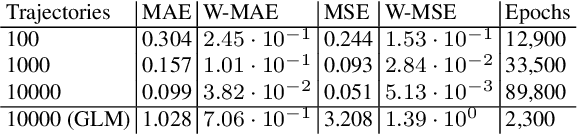
Continuous-time Markov chains are used to model stochastic systems where transitions can occur at irregular times, e.g., birth-death processes, chemical reaction networks, population dynamics, and gene regulatory networks. We develop a method to learn a continuous-time Markov chain's transition rate functions from fully observed time series. In contrast with existing methods, our method allows for transition rates to depend nonlinearly on both state variables and external covariates. The Gillespie algorithm is used to generate trajectories of stochastic systems where propensity functions (reaction rates) are known. Our method can be viewed as the inverse: given trajectories of a stochastic reaction network, we generate estimates of the propensity functions. While previous methods used linear or log-linear methods to link transition rates to covariates, we use neural networks, increasing the capacity and potential accuracy of learned models. In the chemical context, this enables the method to learn propensity functions from non-mass-action kinetics. We test our method with synthetic data generated from a variety of systems with known transition rates. We show that our method learns these transition rates with considerably more accuracy than log-linear methods, in terms of mean absolute error between ground truth and predicted transition rates. We also demonstrate an application of our methods to open-loop control of a continuous-time Markov chain.
Drift Identification for Lévy alpha-Stable Stochastic Systems
Dec 06, 2022



This paper focuses on a stochastic system identification problem: given time series observations of a stochastic differential equation (SDE) driven by L\'{e}vy $\alpha$-stable noise, estimate the SDE's drift field. For $\alpha$ in the interval $[1,2)$, the noise is heavy-tailed, leading to computational difficulties for methods that compute transition densities and/or likelihoods in physical space. We propose a Fourier space approach that centers on computing time-dependent characteristic functions, i.e., Fourier transforms of time-dependent densities. Parameterizing the unknown drift field using Fourier series, we formulate a loss consisting of the squared error between predicted and empirical characteristic functions. We minimize this loss with gradients computed via the adjoint method. For a variety of one- and two-dimensional problems, we demonstrate that this method is capable of learning drift fields in qualitative and/or quantitative agreement with ground truth fields.
Dynamic Learning of Correlation Potentials for a Time-Dependent Kohn-Sham System
Dec 14, 2021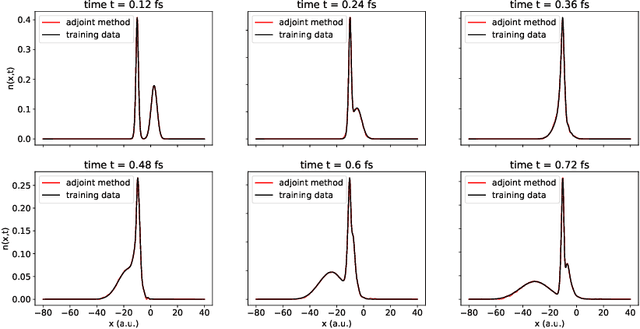
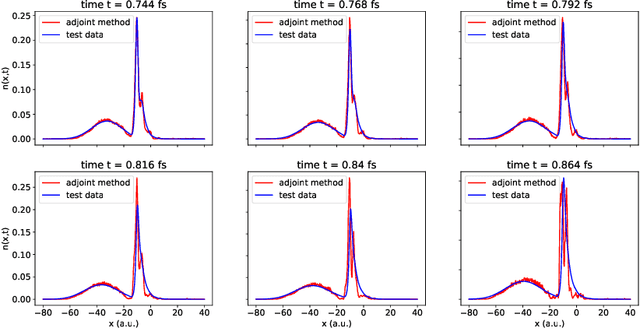

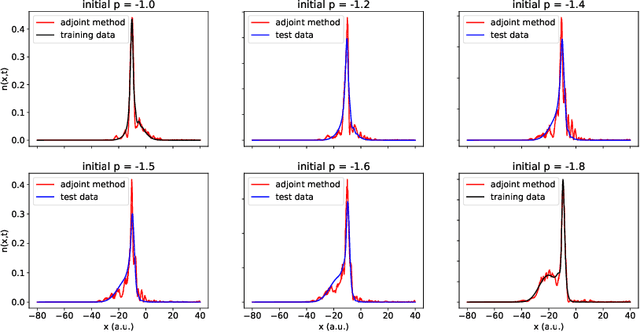
We develop methods to learn the correlation potential for a time-dependent Kohn-Sham (TDKS) system in one spatial dimension. We start from a low-dimensional two-electron system for which we can numerically solve the time-dependent Schr\"odinger equation; this yields electron densities suitable for training models of the correlation potential. We frame the learning problem as one of optimizing a least-squares objective subject to the constraint that the dynamics obey the TDKS equation. Applying adjoints, we develop efficient methods to compute gradients and thereby learn models of the correlation potential. Our results show that it is possible to learn values of the correlation potential such that the resulting electron densities match ground truth densities. We also show how to learn correlation potential functionals with memory, demonstrating one such model that yields reasonable results for trajectories outside the training set.
Equity-Directed Bootstrapping: Examples and Analysis
Aug 14, 2021
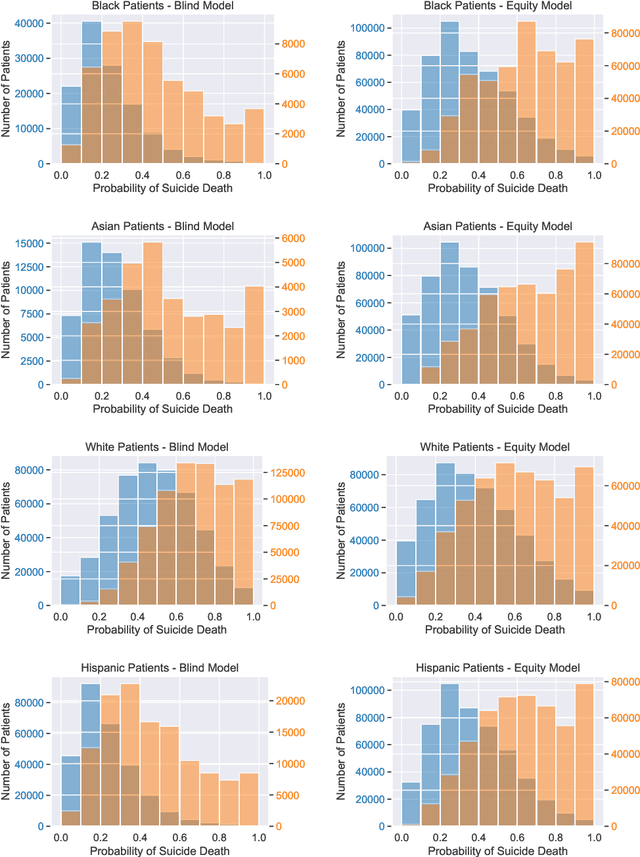
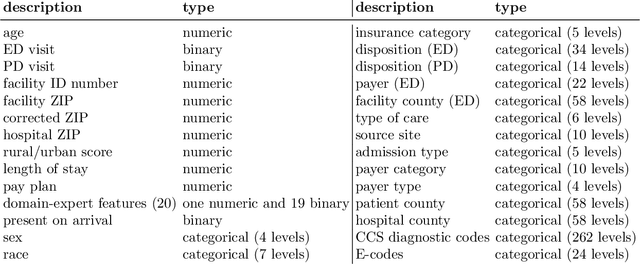
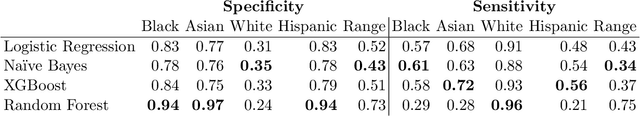
When faced with severely imbalanced binary classification problems, we often train models on bootstrapped data in which the number of instances of each class occur in a more favorable ratio, e.g., one. We view algorithmic inequity through the lens of imbalanced classification: in order to balance the performance of a classifier across groups, we can bootstrap to achieve training sets that are balanced with respect to both labels and group identity. For an example problem with severe class imbalance---prediction of suicide death from administrative patient records---we illustrate how an equity-directed bootstrap can bring test set sensitivities and specificities much closer to satisfying the equal odds criterion. In the context of na\"ive Bayes and logistic regression, we analyze the equity-directed bootstrap, demonstrating that it works by bringing odds ratios close to one, and linking it to methods involving intercept adjustment, thresholding, and weighting.
Statistical learning method for predicting density-matrix based electron dynamics
Jul 31, 2021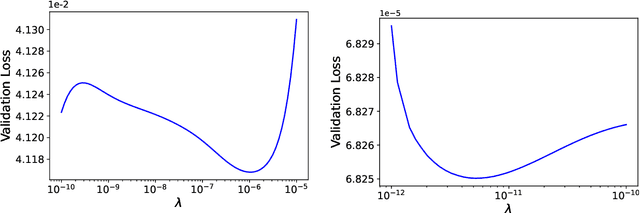

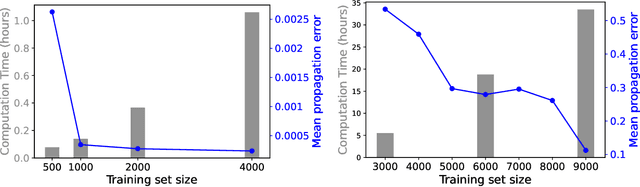
We develop a statistical method to learn a molecular Hamiltonian matrix from a time-series of electron density matrices. We extend our previous method to larger molecular systems by incorporating physical properties to reduce dimensionality, while also exploiting regularization techniques like ridge regression for addressing multicollinearity. With the learned Hamiltonian we can solve the Time-Dependent Hartree-Fock (TDHF) equation to propagate the electron density in time, and predict its dynamics for field-free and field-on scenarios. We observe close quantitative agreement between the predicted dynamics and ground truth for both field-off trajectories similar to the training data, and field-on trajectories outside of the training data.
Estimating Vector Fields from Noisy Time Series
Dec 06, 2020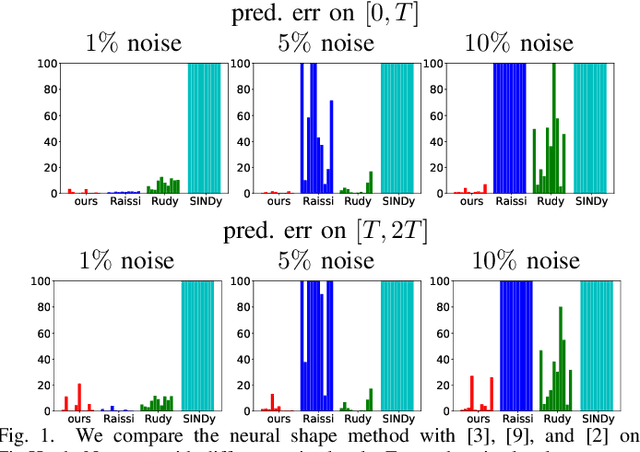
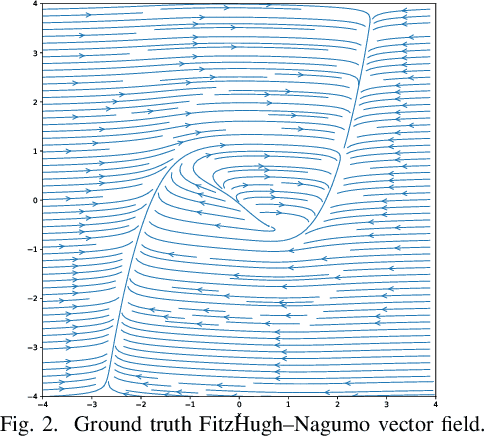
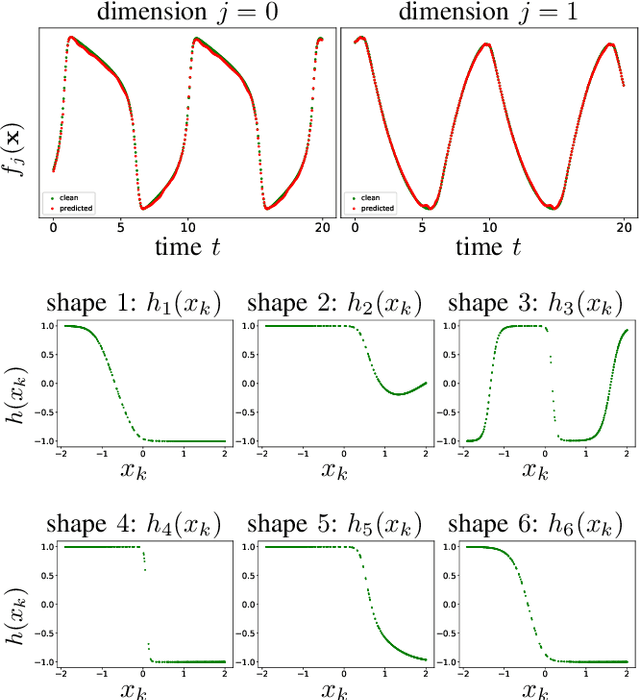
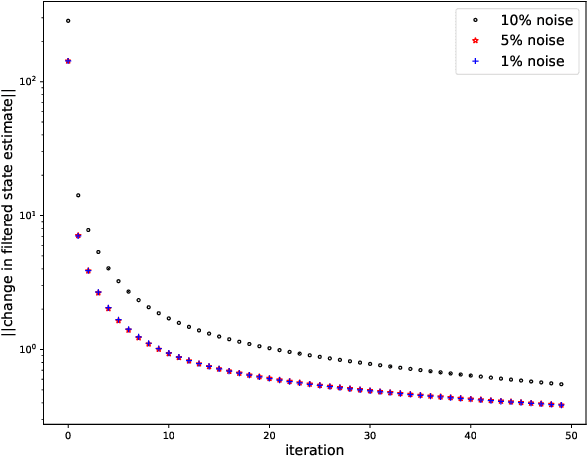
While there has been a surge of recent interest in learning differential equation models from time series, methods in this area typically cannot cope with highly noisy data. We break this problem into two parts: (i) approximating the unknown vector field (or right-hand side) of the differential equation, and (ii) dealing with noise. To deal with (i), we describe a neural network architecture consisting of tensor products of one-dimensional neural shape functions. For (ii), we propose an alternating minimization scheme that switches between vector field training and filtering steps, together with multiple trajectories of training data. We find that the neural shape function architecture retains the approximation properties of dense neural networks, enables effective computation of vector field error, and allows for graphical interpretability, all for data/systems in any finite dimension $d$. We also study the combination of either our neural shape function method or existing differential equation learning methods with alternating minimization and multiple trajectories. We find that retrofitting any learning method in this way boosts the method's robustness to noise. While in their raw form the methods struggle with 1% Gaussian noise, after retrofitting, they learn accurate vector fields from data with 10% Gaussian noise.
A non-autonomous equation discovery method for time signal classification
Nov 22, 2020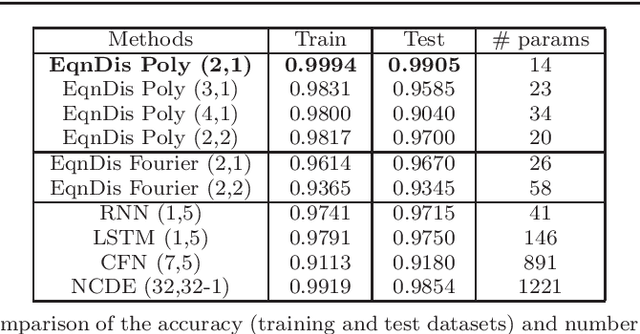
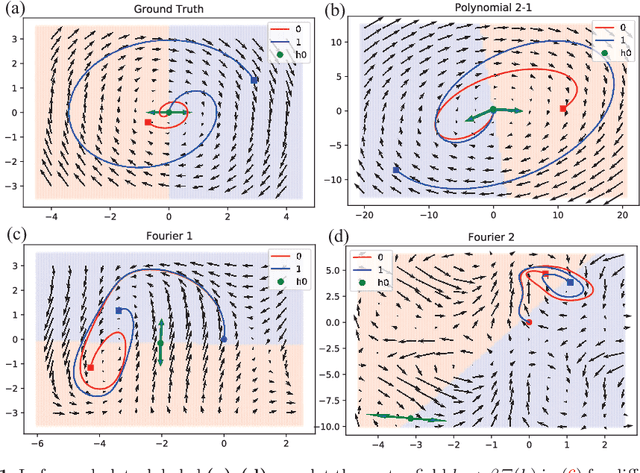
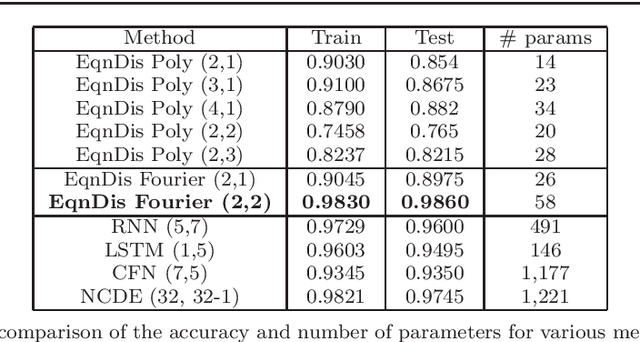
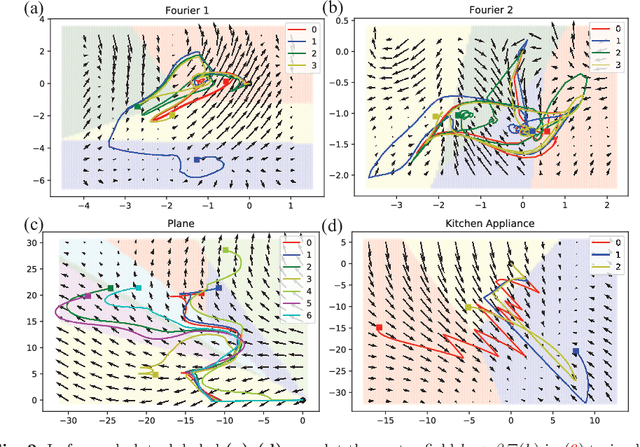
Certain neural network architectures, in the infinite-layer limit, lead to systems of nonlinear differential equations. Motivated by this idea, we develop a framework for analyzing time signals based on non-autonomous dynamical equations. We view the time signal as a forcing function for a dynamical system that governs a time-evolving hidden variable. As in equation discovery, the dynamical system is represented using a dictionary of functions and the coefficients are learned from data. This framework is applied to the time signal classification problem. We show how gradients can be efficiently computed using the adjoint method, and we apply methods from dynamical systems to establish stability of the classifier. Through a variety of experiments, on both synthetic and real datasets, we show that the proposed method uses orders of magnitude fewer parameters than competing methods, while achieving comparable accuracy. We created the synthetic datasets using dynamical systems of increasing complexity; though the ground truth vector fields are often polynomials, we find consistently that a Fourier dictionary yields the best results. We also demonstrate how the proposed method yields graphical interpretability in the form of phase portraits.