 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNonlinear Optimal Control of Electron Dynamics within Hartree-Fock Theory
Dec 04, 2024Consider the problem of determining the optimal applied electric field to drive a molecule from an initial state to a desired target state. For even moderately sized molecules, solving this problem directly using the exact equations of motion -- the time-dependent Schr\"odinger equation (TDSE) -- is numerically intractable. We present a solution of this problem within time-dependent Hartree-Fock (TDHF) theory, a mean field approximation of the TDSE. Optimality is defined in terms of minimizing the total control effort while maximizing the overlap between desired and achieved target states. We frame this problem as an optimization problem constrained by the nonlinear TDHF equations; we solve it using trust region optimization with gradients computed via a custom-built adjoint state method. For three molecular systems, we show that with very small neural network parametrizations of the control, our method yields solutions that achieve desired targets within acceptable constraints and tolerances.
Scalable learning of potentials to predict time-dependent Hartree-Fock dynamics
Aug 08, 2024We propose a framework to learn the time-dependent Hartree-Fock (TDHF) inter-electronic potential of a molecule from its electron density dynamics. Though the entire TDHF Hamiltonian, including the inter-electronic potential, can be computed from first principles, we use this problem as a testbed to develop strategies that can be applied to learn \emph{a priori} unknown terms that arise in other methods/approaches to quantum dynamics, e.g., emerging problems such as learning exchange-correlation potentials for time-dependent density functional theory. We develop, train, and test three models of the TDHF inter-electronic potential, each parameterized by a four-index tensor of size up to $60 \times 60 \times 60 \times 60$. Two of the models preserve Hermitian symmetry, while one model preserves an eight-fold permutation symmetry that implies Hermitian symmetry. Across seven different molecular systems, we find that accounting for the deeper eight-fold symmetry leads to the best-performing model across three metrics: training efficiency, test set predictive power, and direct comparison of true and learned inter-electronic potentials. All three models, when trained on ensembles of field-free trajectories, generate accurate electron dynamics predictions even in a field-on regime that lies outside the training set. To enable our models to scale to large molecular systems, we derive expressions for Jacobian-vector products that enable iterative, matrix-free training.
Dynamic Learning of Correlation Potentials for a Time-Dependent Kohn-Sham System
Dec 14, 2021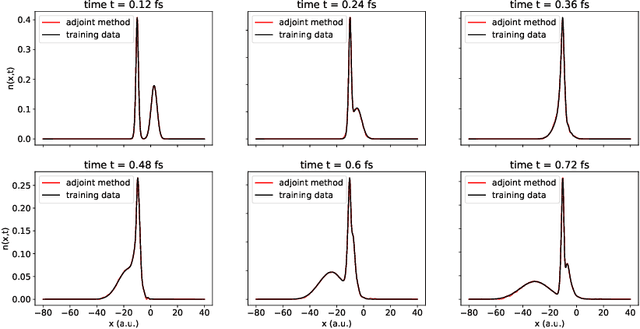
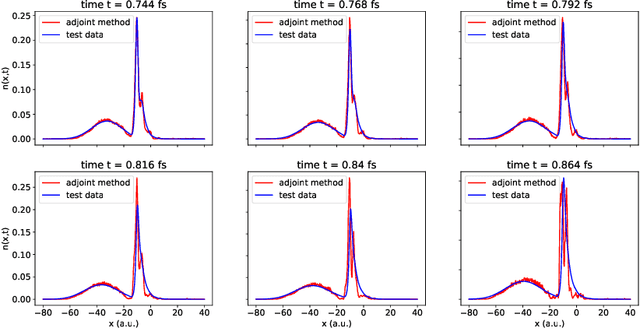

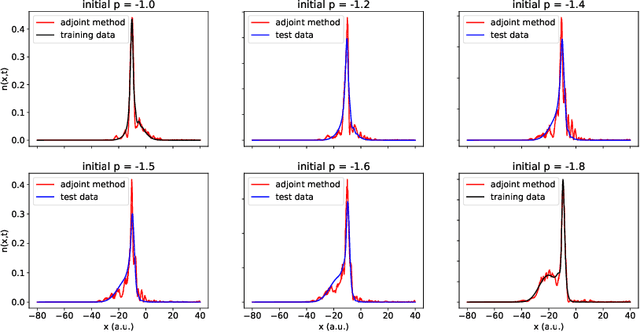
We develop methods to learn the correlation potential for a time-dependent Kohn-Sham (TDKS) system in one spatial dimension. We start from a low-dimensional two-electron system for which we can numerically solve the time-dependent Schr\"odinger equation; this yields electron densities suitable for training models of the correlation potential. We frame the learning problem as one of optimizing a least-squares objective subject to the constraint that the dynamics obey the TDKS equation. Applying adjoints, we develop efficient methods to compute gradients and thereby learn models of the correlation potential. Our results show that it is possible to learn values of the correlation potential such that the resulting electron densities match ground truth densities. We also show how to learn correlation potential functionals with memory, demonstrating one such model that yields reasonable results for trajectories outside the training set.
Statistical learning method for predicting density-matrix based electron dynamics
Jul 31, 2021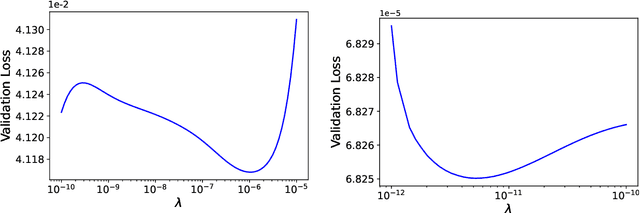

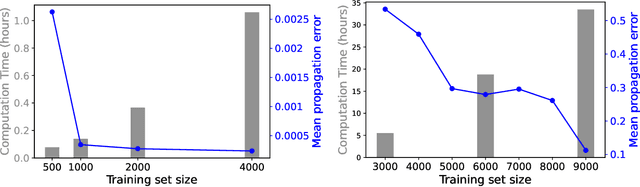
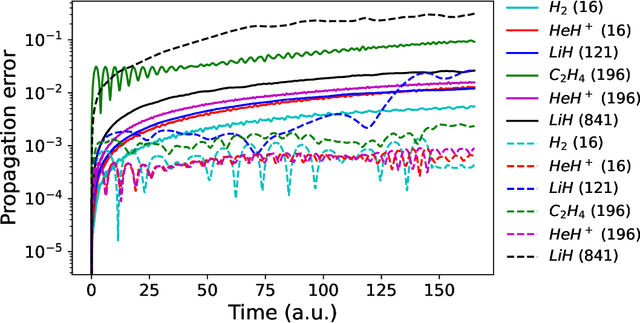
We develop a statistical method to learn a molecular Hamiltonian matrix from a time-series of electron density matrices. We extend our previous method to larger molecular systems by incorporating physical properties to reduce dimensionality, while also exploiting regularization techniques like ridge regression for addressing multicollinearity. With the learned Hamiltonian we can solve the Time-Dependent Hartree-Fock (TDHF) equation to propagate the electron density in time, and predict its dynamics for field-free and field-on scenarios. We observe close quantitative agreement between the predicted dynamics and ground truth for both field-off trajectories similar to the training data, and field-on trajectories outside of the training data.
Machine Learning a Molecular Hamiltonian for Predicting Electron Dynamics
Aug 31, 2020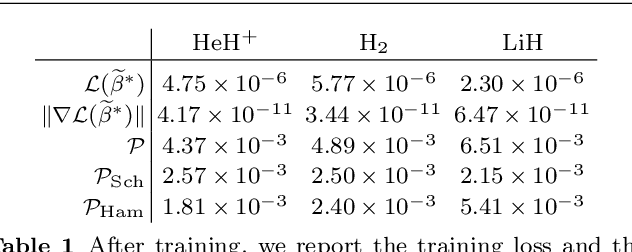
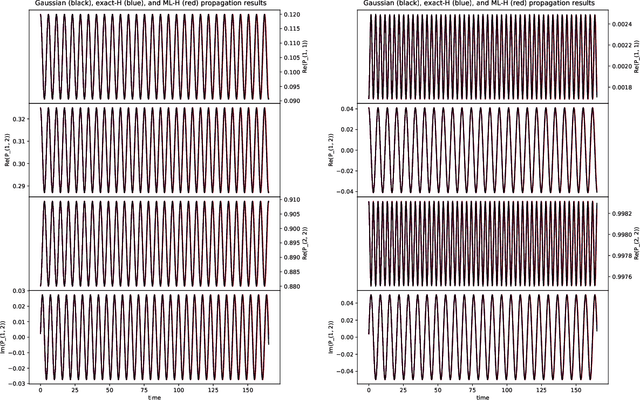
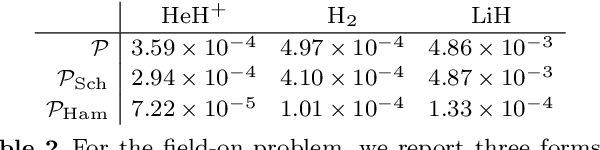
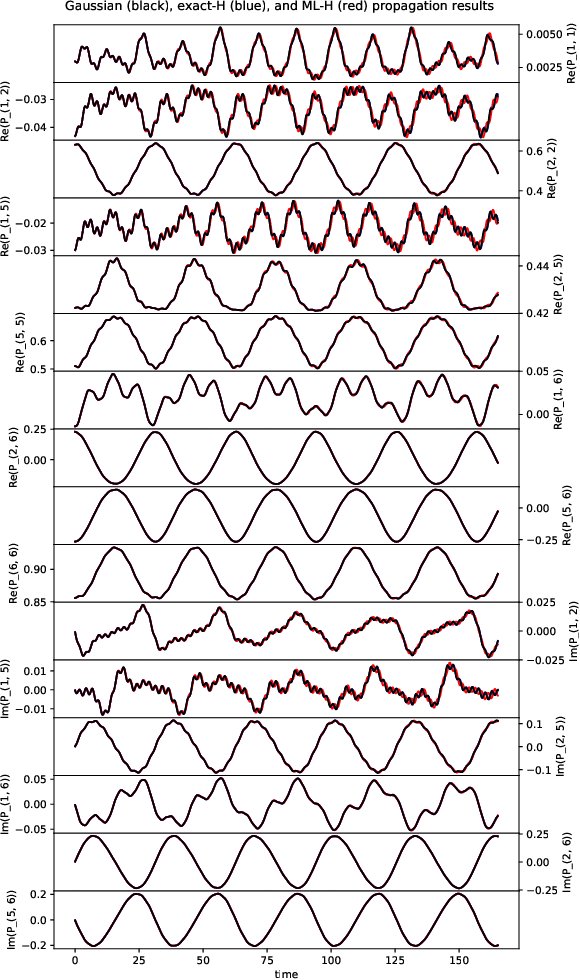
We develop a computational method to learn a molecular Hamiltonian matrix from matrix-valued time series of the electron density. As we demonstrate for three small molecules, the resulting Hamiltonians can be used for electron density evolution, producing highly accurate results even when propagating 1000 time steps beyond the training data. As a more rigorous test, we use the learned Hamiltonians to simulate electron dynamics in the presence of an applied electric field, extrapolating to a problem that is beyond the field-free training data. We find that the resulting electron dynamics predicted by our learned Hamiltonian are in close quantitative agreement with the ground truth. Our method relies on combining a reduced-dimensional, linear statistical model of the Hamiltonian with a time-discretization of the quantum Liouville equation within time-dependent Hartree Fock theory. We train the model using a least-squares solver, avoiding numerous, CPU-intensive optimization steps. For both field-free and field-on problems, we quantify training and propagation errors, highlighting areas for future development.