 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTASTE: A Designer-Annotated Multi-Dimensional Preference Dataset for AI-Generated Graphic Design
May 20, 2026Text-to-image models produce graphic design at production scale, but their supervision comes from photo-style preference data with a single overall verdict per comparison. Designers evaluate along several distinct axes, including typography, visual hierarchy, color harmony, layout, and brief fidelity, and a single label collapses them. We release TASTE (Typography, Aesthetics, Spatial, Tone, Etc.): ten professional designers ranked outputs from four current text-to-image models on nine criteria across two disjoint cohorts, yielding 1,600 ratings per criterion plus per-image hallucination flags on the holistic-preference cohorts. We pair the dataset with three contributions. First, a criterion-agnostic signal test framework, using Kendall's tau, majority probability, and Condorcet cycles against exact iid-uniform nulls at p = 4 and R = 5, places designer agreement on graphic design between food and movie preferences and photo-style image quality, with every TASTE criterion rejecting the random-rater null. Second, no pre-trained system in our benchmark, including six open-weight VLM judges from 3B to 33B parameters and three dedicated T2I scorers, HPSv2.1, PickScore-v1, and LAION-Aesthetic-V2, exceeds 0.55 macro agreement with the 5-designer majority; VLM judges trade off position bias against content sensitivity, so scaling moves along this frontier without improving accuracy. Third, a small pairwise-difference head trained on TASTE reaches 0.611, closing roughly half the gap to the 0.741 single-rater ceiling.
Structural Evaluation Metrics for SVG Generation via Leave-One-Out Analysis
Apr 09, 2026Scalable Vector Graphics (SVG) represent visual content as structured, editable code. Each element (path, shape, or text node) can be individually inspected, transformed, or removed. This structural editability is a main motivation for SVG generation, yet prevailing evaluation protocols primarily reduce the output to a single similarity score against a reference image or input texts, measuring how faithfully the result reproduces an image or follows the instructions, but not how well it preserves the structural properties that make SVG valuable. In particular, existing metrics cannot determine which generated elements contribute positively to overall visual quality, how visual concepts map to specific parts of the code, or whether the generated output supports meaningful downstream editing. We introduce element-level leave-one-out (LOO) analysis, inspired by the classic jackknife estimator. The procedure renders the SVG with and without each element, measures the resulting visual change, and derives a suite of structural quality metrics. Despite its simplicity, the jackknife's capacity to decompose an aggregate statistic into per-sample contributions translates directly to this setting. From a single mechanism, we obtain: (1) quality scores per element through LOO scoring that enable zero-shot artifact detection; (2) concept-element attribution that maps each element to the visual concept it serves; and (3) four structural metrics, purity, coverage, compactness, and locality, that quantify SVG modularity from complementary perspectives. We validate these metrics on over 19,000 edits (5 types) across 5 generation systems and 3 complexity tiers.
Graphic-Design-Bench: A Comprehensive Benchmark for Evaluating AI on Graphic Design Tasks
Apr 07, 2026We introduce GraphicDesignBench (GDB), the first comprehensive benchmark suite designed specifically to evaluate AI models on the full breadth of professional graphic design tasks. Unlike existing benchmarks that focus on natural-image understanding or generic text-to-image synthesis, GDB targets the unique challenges of professional design work: translating communicative intent into structured layouts, rendering typographically faithful text, manipulating layered compositions, producing valid vector graphics, and reasoning about animation. The suite comprises 50 tasks organized along five axes: layout, typography, infographics, template & design semantics and animation, each evaluated under both understanding and generation settings, and grounded in real-world design templates drawn from the LICA layered-composition dataset. We evaluate a set of frontier closed-source models using a standardized metric taxonomy covering spatial accuracy, perceptual quality, text fidelity, semantic alignment, and structural validity. Our results reveal that current models fall short on the core challenges of professional design: spatial reasoning over complex layouts, faithful vector code generation, fine-grained typographic perception, and temporal decomposition of animations remain largely unsolved. While high-level semantic understanding is within reach, the gap widens sharply as tasks demand precision, structure, and compositional awareness. GDB provides a rigorous, reproducible testbed for tracking progress toward AI systems that can function as capable design collaborators. The full evaluation framework is publicly available.
Hierarchical Sparse Bayesian Multitask Model with Scalable Inference for Microbiome Analysis
Feb 04, 2025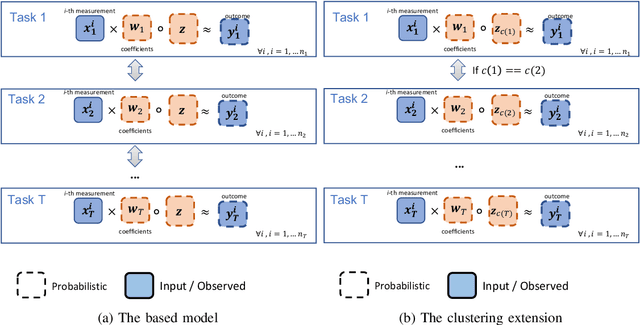
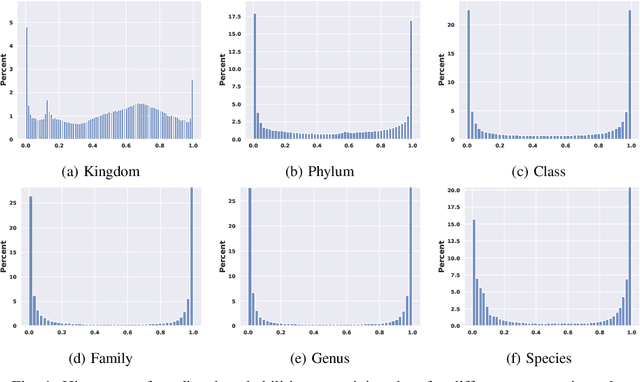
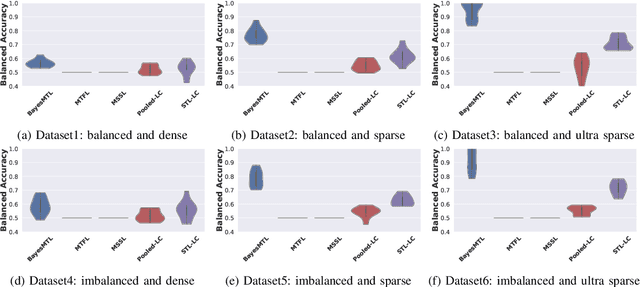
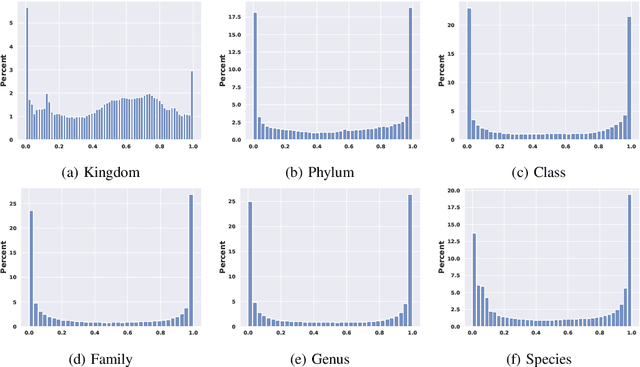
This paper proposes a hierarchical Bayesian multitask learning model that is applicable to the general multi-task binary classification learning problem where the model assumes a shared sparsity structure across different tasks. We derive a computationally efficient inference algorithm based on variational inference to approximate the posterior distribution. We demonstrate the potential of the new approach on various synthetic datasets and for predicting human health status based on microbiome profile. Our analysis incorporates data pooled from multiple microbiome studies, along with a comprehensive comparison with other benchmark methods. Results in synthetic datasets show that the proposed approach has superior support recovery property when the underlying regression coefficients share a common sparsity structure across different tasks. Our experiments on microbiome classification demonstrate the utility of the method in extracting informative taxa while providing well-calibrated predictions with uncertainty quantification and achieving competitive performance in terms of prediction metrics. Notably, despite the heterogeneity of the pooled datasets (e.g., different experimental objectives, laboratory setups, sequencing equipment, patient demographics), our method delivers robust results.
Deep Active Learning based Experimental Design to Uncover Synergistic Genetic Interactions for Host Targeted Therapeutics
Feb 03, 2025



Recent technological advances have introduced new high-throughput methods for studying host-virus interactions, but testing synergistic interactions between host gene pairs during infection remains relatively slow and labor intensive. Identification of multiple gene knockdowns that effectively inhibit viral replication requires a search over the combinatorial space of all possible target gene pairs and is infeasible via brute-force experiments. Although active learning methods for sequential experimental design have shown promise, existing approaches have generally been restricted to single-gene knockdowns or small-scale double knockdown datasets. In this study, we present an integrated Deep Active Learning (DeepAL) framework that incorporates information from a biological knowledge graph (SPOKE, the Scalable Precision Medicine Open Knowledge Engine) to efficiently search the configuration space of a large dataset of all pairwise knockdowns of 356 human genes in HIV infection. Through graph representation learning, the framework is able to generate task-specific representations of genes while also balancing the exploration-exploitation trade-off to pinpoint highly effective double-knockdown pairs. We additionally present an ensemble method for uncertainty quantification and an interpretation of the gene pairs selected by our algorithm via pathway analysis. To our knowledge, this is the first work to show promising results on double-gene knockdown experimental data of appreciable scale (356 by 356 matrix).
A Graphical Model for Fusing Diverse Microbiome Data
Aug 21, 2022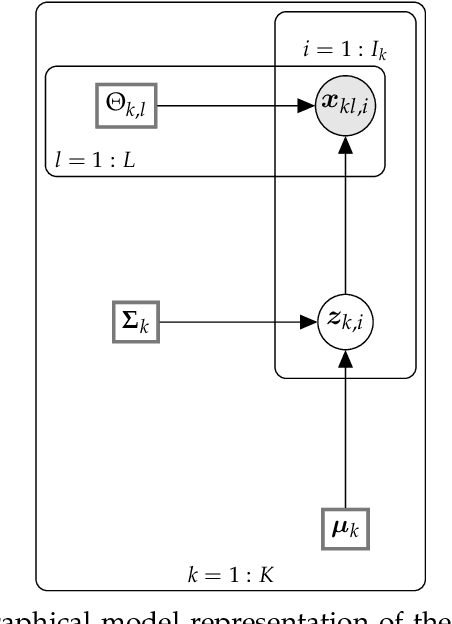
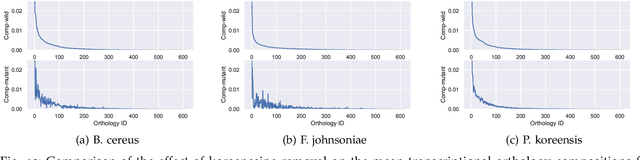
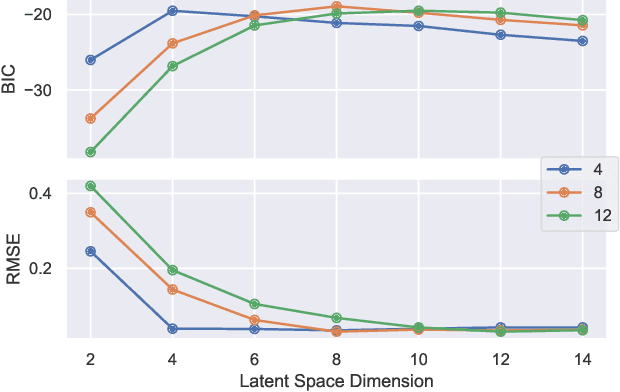
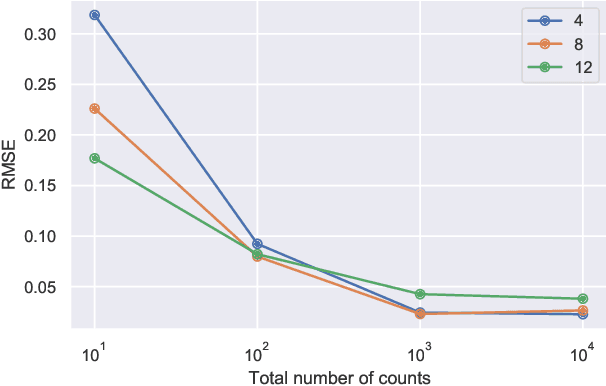
This paper develops a Bayesian graphical model for fusing disparate types of count data. The motivating application is the study of bacterial communities from diverse high dimensional features, in this case transcripts, collected from different treatments. In such datasets, there are no explicit correspondences between the communities and each correspond to different factors, making data fusion challenging. We introduce a flexible multinomial-Gaussian generative model for jointly modeling such count data. This latent variable model jointly characterizes the observed data through a common multivariate Gaussian latent space that parameterizes the set of multinomial probabilities of the transcriptome counts. The covariance matrix of the latent variables induces a covariance matrix of co-dependencies between all the transcripts, effectively fusing multiple data sources. We present a computationally scalable variational Expectation-Maximization (EM) algorithm for inferring the latent variables and the parameters of the model. The inferred latent variables provide a common dimensionality reduction for visualizing the data and the inferred parameters provide a predictive posterior distribution. In addition to simulation studies that demonstrate the variational EM procedure, we apply our model to a bacterial microbiome dataset.