 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHOP: Heterogeneous Topology-based Multimodal Entanglement for Co-Speech Gesture Generation
Mar 03, 2025



Co-speech gestures are crucial non-verbal cues that enhance speech clarity and expressiveness in human communication, which have attracted increasing attention in multimodal research. While the existing methods have made strides in gesture accuracy, challenges remain in generating diverse and coherent gestures, as most approaches assume independence among multimodal inputs and lack explicit modeling of their interactions. In this work, we propose a novel multimodal learning method named HOP for co-speech gesture generation that captures the heterogeneous entanglement between gesture motion, audio rhythm, and text semantics, enabling the generation of coordinated gestures. By leveraging spatiotemporal graph modeling, we achieve the alignment of audio and action. Moreover, to enhance modality coherence, we build the audio-text semantic representation based on a reprogramming module, which is beneficial for cross-modality adaptation. Our approach enables the trimodal system to learn each other's features and represent them in the form of topological entanglement. Extensive experiments demonstrate that HOP achieves state-of-the-art performance, offering more natural and expressive co-speech gesture generation. More information, codes, and demos are available here: https://star-uu-wang.github.io/HOP/
LLM-Powered Explanations: Unraveling Recommendations Through Subgraph Reasoning
Jun 22, 2024



Recommender systems are pivotal in enhancing user experiences across various web applications by analyzing the complicated relationships between users and items. Knowledge graphs(KGs) have been widely used to enhance the performance of recommender systems. However, KGs are known to be noisy and incomplete, which are hard to provide reliable explanations for recommendation results. An explainable recommender system is crucial for the product development and subsequent decision-making. To address these challenges, we introduce a novel recommender that synergies Large Language Models (LLMs) and KGs to enhance the recommendation and provide interpretable results. Specifically, we first harness the power of LLMs to augment KG reconstruction. LLMs comprehend and decompose user reviews into new triples that are added into KG. In this way, we can enrich KGs with explainable paths that express user preferences. To enhance the recommendation on augmented KGs, we introduce a novel subgraph reasoning module that effectively measures the importance of nodes and discovers reasoning for recommendation. Finally, these reasoning paths are fed into the LLMs to generate interpretable explanations of the recommendation results. Our approach significantly enhances both the effectiveness and interpretability of recommender systems, especially in cross-selling scenarios where traditional methods falter. The effectiveness of our approach has been rigorously tested on four open real-world datasets, with our methods demonstrating a superior performance over contemporary state-of-the-art techniques by an average improvement of 12%. The application of our model in a multinational engineering and technology company cross-selling recommendation system further underscores its practical utility and potential to redefine recommendation practices through improved accuracy and user trust.
Contrastive Dual-Interaction Graph Neural Network for Molecular Property Prediction
May 04, 2024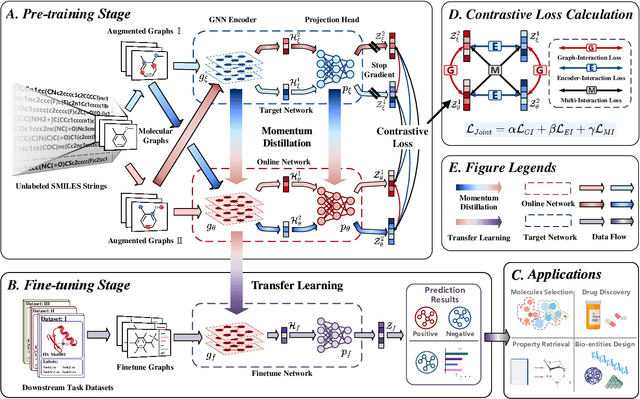
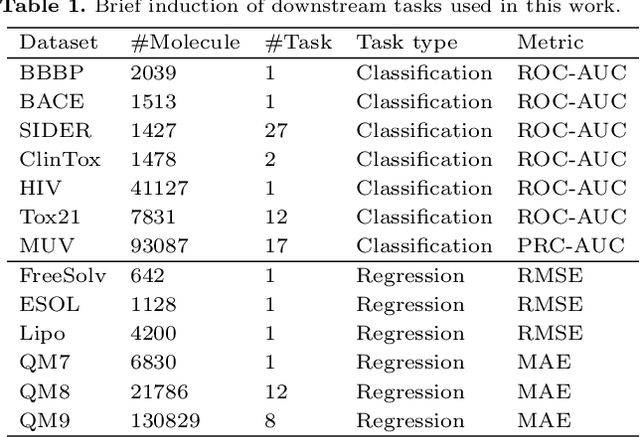
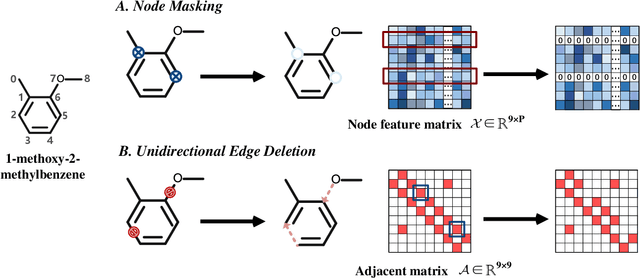
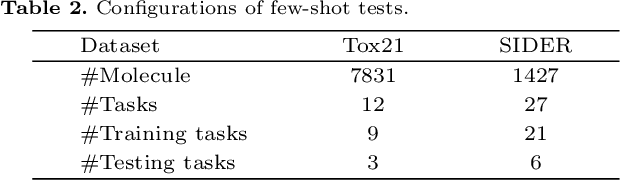
Molecular property prediction is a key component of AI-driven drug discovery and molecular characterization learning. Despite recent advances, existing methods still face challenges such as limited ability to generalize, and inadequate representation of learning from unlabeled data, especially for tasks specific to molecular structures. To address these limitations, we introduce DIG-Mol, a novel self-supervised graph neural network framework for molecular property prediction. This architecture leverages the power of contrast learning with dual interaction mechanisms and unique molecular graph enhancement strategies. DIG-Mol integrates a momentum distillation network with two interconnected networks to efficiently improve molecular characterization. The framework's ability to extract key information about molecular structure and higher-order semantics is supported by minimizing loss of contrast. We have established DIG-Mol's state-of-the-art performance through extensive experimental evaluation in a variety of molecular property prediction tasks. In addition to demonstrating superior transferability in a small number of learning scenarios, our visualizations highlight DIG-Mol's enhanced interpretability and representation capabilities. These findings confirm the effectiveness of our approach in overcoming challenges faced by traditional methods and mark a significant advance in molecular property prediction.
Towards Complex Dynamic Physics System Simulation with Graph Neural ODEs
May 25, 2023



The great learning ability of deep learning models facilitates us to comprehend the real physical world, making learning to simulate complicated particle systems a promising endeavour. However, the complex laws of the physical world pose significant challenges to the learning based simulations, such as the varying spatial dependencies between interacting particles and varying temporal dependencies between particle system states in different time stamps, which dominate particles' interacting behaviour and the physical systems' evolution patterns. Existing learning based simulation methods fail to fully account for the complexities, making them unable to yield satisfactory simulations. To better comprehend the complex physical laws, this paper proposes a novel learning based simulation model- Graph Networks with Spatial-Temporal neural Ordinary Equations (GNSTODE)- that characterizes the varying spatial and temporal dependencies in particle systems using a united end-to-end framework. Through training with real-world particle-particle interaction observations, GNSTODE is able to simulate any possible particle systems with high precisions. We empirically evaluate GNSTODE's simulation performance on two real-world particle systems, Gravity and Coulomb, with varying levels of spatial and temporal dependencies. The results show that the proposed GNSTODE yields significantly better simulations than state-of-the-art learning based simulation methods, which proves that GNSTODE can serve as an effective solution to particle simulations in real-world application.
How Expressive are Spectral-Temporal Graph Neural Networks for Time Series Forecasting?
May 11, 2023


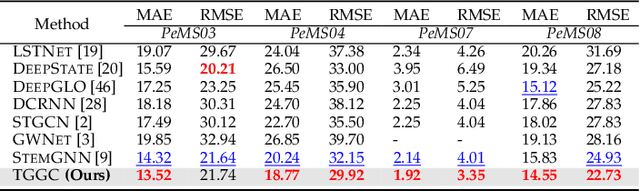
Spectral-temporal graph neural network is a promising abstraction underlying most time series forecasting models that are based on graph neural networks (GNNs). However, more is needed to know about the underpinnings of this branch of methods. In this paper, we establish a theoretical framework that unravels the expressive power of spectral-temporal GNNs. Our results show that linear spectral-temporal GNNs are universal under mild assumptions, and their expressive power is bounded by our extended first-order Weisfeiler-Leman algorithm on discrete-time dynamic graphs. To make our findings useful in practice on valid instantiations, we discuss related constraints in detail and outline a theoretical blueprint for designing spatial and temporal modules in spectral domains. Building on these insights and to demonstrate how powerful spectral-temporal GNNs are based on our framework, we propose a simple instantiation named Temporal Graph GegenConv (TGC), which significantly outperforms most existing models with only linear components and shows better model efficiency.