 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHOP: Heterogeneous Topology-based Multimodal Entanglement for Co-Speech Gesture Generation
Mar 03, 2025



Co-speech gestures are crucial non-verbal cues that enhance speech clarity and expressiveness in human communication, which have attracted increasing attention in multimodal research. While the existing methods have made strides in gesture accuracy, challenges remain in generating diverse and coherent gestures, as most approaches assume independence among multimodal inputs and lack explicit modeling of their interactions. In this work, we propose a novel multimodal learning method named HOP for co-speech gesture generation that captures the heterogeneous entanglement between gesture motion, audio rhythm, and text semantics, enabling the generation of coordinated gestures. By leveraging spatiotemporal graph modeling, we achieve the alignment of audio and action. Moreover, to enhance modality coherence, we build the audio-text semantic representation based on a reprogramming module, which is beneficial for cross-modality adaptation. Our approach enables the trimodal system to learn each other's features and represent them in the form of topological entanglement. Extensive experiments demonstrate that HOP achieves state-of-the-art performance, offering more natural and expressive co-speech gesture generation. More information, codes, and demos are available here: https://star-uu-wang.github.io/HOP/
Contrastive Dual-Interaction Graph Neural Network for Molecular Property Prediction
May 04, 2024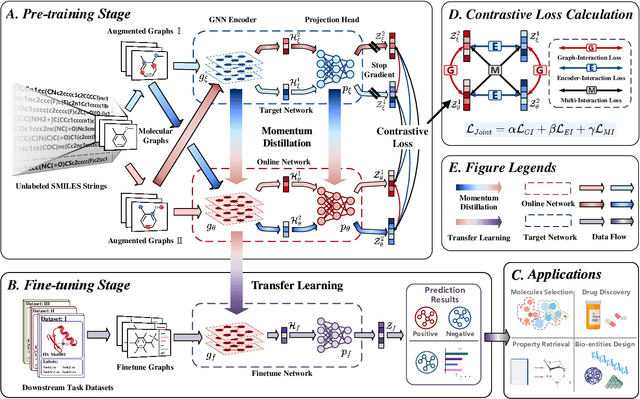
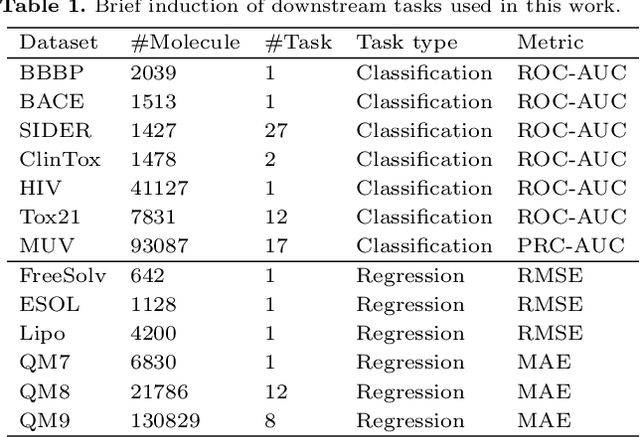
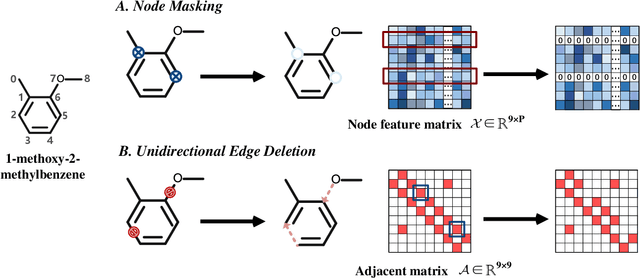
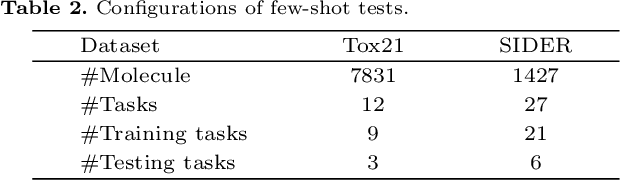
Molecular property prediction is a key component of AI-driven drug discovery and molecular characterization learning. Despite recent advances, existing methods still face challenges such as limited ability to generalize, and inadequate representation of learning from unlabeled data, especially for tasks specific to molecular structures. To address these limitations, we introduce DIG-Mol, a novel self-supervised graph neural network framework for molecular property prediction. This architecture leverages the power of contrast learning with dual interaction mechanisms and unique molecular graph enhancement strategies. DIG-Mol integrates a momentum distillation network with two interconnected networks to efficiently improve molecular characterization. The framework's ability to extract key information about molecular structure and higher-order semantics is supported by minimizing loss of contrast. We have established DIG-Mol's state-of-the-art performance through extensive experimental evaluation in a variety of molecular property prediction tasks. In addition to demonstrating superior transferability in a small number of learning scenarios, our visualizations highlight DIG-Mol's enhanced interpretability and representation capabilities. These findings confirm the effectiveness of our approach in overcoming challenges faced by traditional methods and mark a significant advance in molecular property prediction.